ARTICLES
Continuing oversight through site monitoring: experiences of an institutional ethics committee in an Indian tertiary-care hospital
Yashashri C Shetty, Padmaja Marathe, Sandhya Kamat, Urmila Thatte
DOI: https://doi.org/10.20529/IJME.2012.006
Abstract
WHO-TDR and the Indian Council of Medical Research recommend site visits by institutional ethics committees (IECs) for continued oversight, to ensure the ethical conduct of research. Our IEC conducted seven site visits in 2008-2009 using a standardised format to monitor adherence to protocol and the informed consent process.
The study identified issues related to informed consent (6, 7), deviation from protocol (5, 7), reporting of study progress to the IEC (3, 7), recruiting additional participants without IEC approval (2, 7), reporting of serious adverse events (1, 7), investigator’s lack of awareness of protocol and the informed consent document (2, 7) and other findings.
Investigators were informed about the findings and were asked to submit an explanation. The IEC issued warnings about not repeating such lapses in the future (5, 7), restricted enrollment of new participants (2, 7), recommended continued good clinical practice training to the study team (4, 7), advised the recruitment of additional study coordinators (2, 7), and requested the submission of adverse event reports (2, 7) or sponsors’ audit reports (2, 7). Our study showed that the ethical conduct of studies can be ensured by conducting routine site monitoring.
Introduction
The continuing review of approved research by institutional ethics committees (IECs) is essential to ensure the ethical conduct of clinical research. IECs perform this duty primarily by reviewing data submitted to them during the conduct of a trial at pre-specified regular intervals. This data includes serious adverse event (SAE) reports, progress reports, reviews of protocol violations, and of amendments of protocol, and related documents submitted by the investigators etc, as recommended under national and international guidelines and legislation (1, 3). This is generally a form of “passive monitoring”. In order to ensure the safety and well being of participants ,as well as to ascertain that potential risks have not altered, these same guidelines also recommend site visits as one of the methods for continuing review by IECs (1, 3).
Site monitoring is a routine activity in the United Kingdom and research ECs carry out proactive monitoring through questionnaires and/or by visiting research sites for pharmaceutical industry-sponsored trials (4). However, IECs in India have neither the mechanisms in place nor the manpower or resources to meet this requirement, and therefore cannot fulfil this obligation (5). Consequently, they rely upon passive monitoring. Additionally, and perhaps as relevant, is the fact that all medical institutions take up investigator-initiated studies where sponsor-driven routine monitoring may not be carried out, emphasising the greater need for continued monitoring by IECs (6).
As a strategy to address this issue, our IEC (located in a tertiary care hospital in India) conducted site monitoring visits according to pre-drafted standard operating procedures (SOPs) (7). Visits to different study sites were organised to monitor the conduct of the ongoing studies. The objectives of these visits were to check compliance of investigators with the protocol and the informed consent process approved by the IEC as well as to assess the level of understanding of the research participants. This paper discusses the observations made during site visits and the subsequent recommendations made to the investigators as also action taken by the IEC.
Methodology
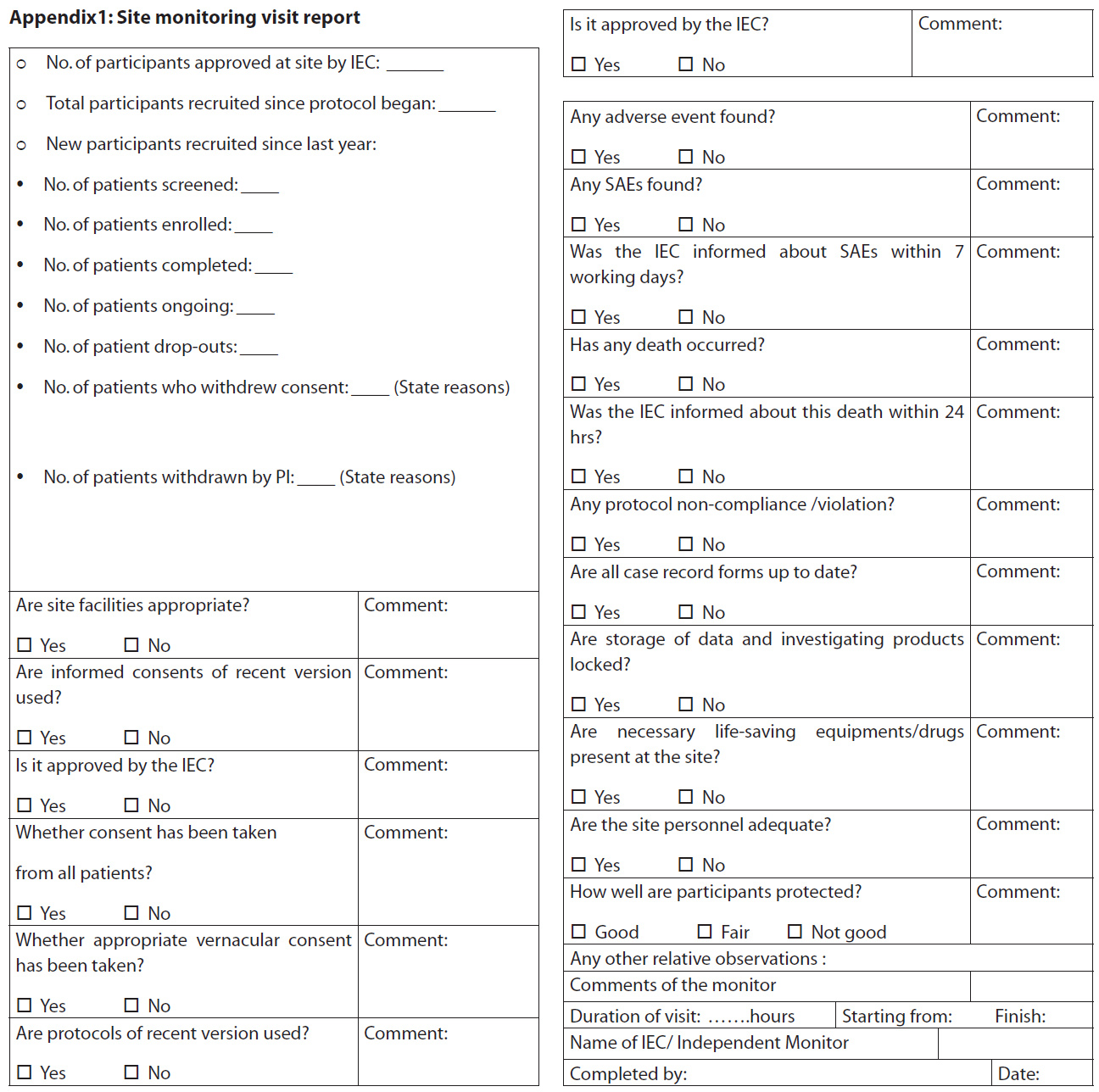
Seven sites were monitored by members of the IEC between January 2008 and December 2010, using a standardised pre-decided format based on the SOPs (Appendix 1). The sites were selected for monitoring either ‘for cause’ (n=5) including incomplete communication from the principal investigator (PI) regarding study progress, a large number of SAEs (deaths) reported from a site, large numbers of protocol deviations, recruitment of additional participants without approval of the IEC and a large number of studies undertaken by the PI at one site or routine (n=2) for investigator-initiated studies.
The site visits were conducted according to the IEC’s SOP number 15 (7). The PIs were informed in writing two weeks in advance about the schedule of site visits, and their acceptance and availability were confirmed before conducting the visits. A team of two IEC members conducted the visits and noted down the observations in the site monitoring report (Appendix 1).The approval of the IEC was obtained to compile and analyse the site visit reports. The reports were analysed for violations and categorised under seven themes: (1) informed consent; (2) deviation from investigational plan , (3) non-reporting of study progress to IEC, (4) IEC approval (5) lack of investigator understanding of protocol and informed consent documents (ICD), (6)SAE reporting, and (7) other findings.
Results
Of the seven studies selected for monitoring, five were pharmaceutical industry-sponsored and two were investigator-initiated studies. All seven were drug trials. The most common findings in these audits were related to the informed consent process (6, 7). Others included deviations from the investigational plan (5, 7), non-reporting of the study’s progress to the IEC (3, 7), recruiting additional participants without IEC approval (2, 7), lack of investigator awareness regarding the protocol and informed consent document (3, 7), and serious adverse event reporting (1, 7). In addition to the above, some other findings were also noted, all of which have been summarised in Table 1.
| Table 1: Violation themes observed during site monitoring by IEC | |
| Violation theme | Monitoring sites (n=7) |
| Informed consent issues | 6/7 |
| Deviation from investigational plan | 5/7 |
| Non-reporting of study progress to IEC | 3/7 |
| Deficiencies in study supervision | 2/7 |
| IEC approval | 2/7 |
| Lack of investigator’s understanding about protocol and informed consent document | 3/7 |
| Serious adverse event reporting | 1/7 |
|
Other findings No source documents found; No coded drugs used; Documents not kept under lock and key; Auditors’ monitoring report missing; PI reported SAE late; Biodata of investigators in the project file not signed |
1/7 [one observation was noted in each of the 7 studies monitored] |
The informed consent process
Six of the seven sites monitored had issues concerning the process of documenting informed consent. Copies of the informed consent documents (ICD) were not available at the site. The explanation given by the investigator was that as it was a collaborative study, the ICDs were archived with the other institution, in spite of the fact that the patients had been recruited from our institution. Other findings included missing signatures of patients (n=1) and principal investigator (n=2). The consent of four participants had been obtained using the vernacular (Hindi) consent form which had not been approved by the IEC, while only the English version had approval. In one study, we found that six participants had been administered the English version of the ICD when they were actually literate in Marathi (local language) and had signed the English consent form in Marathi. The explanation given by the investigator was that the patients’ signatures were obtained only after explaining everything to them. In one case, the ICD was signed 10 days after obtaining the participant’s signature by the PI and co-investigator (Co-I). In one ICD, a nurse of the Institute was used as an impartial witness. In one study, names of all the 30 participants and dates had been filled in by the PI. Without documenting the participant’s consent on the ICD, all the data had been filled in into the case record forms. In one study, informed consent was re-obtained from participants with an amended (approved by IRB) version of the ICD containing information on important new side effects after a delay of 1-3 months. The informed consent addendum was signed by the PI and the Co-I 10 days later, and in the case of 30 participants, a copy of the ICD had not been given to any of them.
In order to assess the participants’ understanding of the study, one participant of each of three studies (a total of three participants) was interviewed. Since the participants were not familiar with the English language, they were interviewed in the local language. It was observed that the participants had a good understanding of the study and the drugs given. They could explain the risks and discomforts of participation in the study. One of the participant’s relatives confided that they were participating in the study as they received free treatment.
Deviations from the investigational plan
At one of the sites, investigators were using an older version of the study protocol. For the investigator-initiated study, participants were not randomised as required in the protocol.
Failure to report study progress to IEC
As per the SOP, our IECs grant approval for a period of one year and then extension of approval after reviewing the annual status report of the study. There was a lapse in submitting the annual status report and taking extension of approval by one of the PIs and 85/125 patients were recruited during the period not covered by valid approval of the IEC.
Recruitment of additional participants without IEC approval
In one of the sponsored studies, the IEC had given permission for recruiting 25 patients while the PI had recruited 30 patients.
Lack of investigator awareness regarding the protocol and informed consent document
At six sites, investigators were interviewed to verify their awareness of the protocol. In one study, the PI did not know the inclusion criteria. At another study site, a ready-reckoner of selection criteria prepared by investigators to facilitate recruitment of participants was found incomplete. This could have led to protocol violations in recruitment.
At three sites, when Co-Is were interviewed in the absence of PIs, it was observed that answers given by Co-Is to questions on inclusion criteria were inappropriate. One Co-I commented that an elaborate consent process would deter patients from participating in the study. In two investigator-initiated studies, the investigators (post-graduate students) were not aware that source documents had to be maintained; patients were not being randomised according to the charts; ICDs that were administered to the participant were not in the language that the patient understood; and participants had not been given a copy of the ICD.
SAE reporting
A total of 20 SAEs from the 1/7 sites were reported to the IEC after seven working days.
Other findings
All the case record forms were found to be incompletely filled in with pencil [25/25] at one site. One of the sites had been monitored by the sponsor, but the monitoring report was missing. The bio-data of the investigators in the project file had not been signed. No source documents were found at two study sites. Documents were not kept under lock and key. A single study co-ordinator was handling multiple studies. At one site, the study had been initiated without having received the approval of the directorate general of foreign trade, India, for shipping of samples abroad.
Action taken by IEC
After monitoring the sites, the teams presented their reports at the next IEC meeting. After the presentation of each report, the findings of monitoring visits were conveyed in writing to the PIs with a request to demonstrate compliance within a specified time. The compliance report was again presented to the IEC and recommendations were given to the PIs. If the PI was found to have deviated from the investigational plan frequently, the IEC put temporary (till PIs gave evidence of taking recommended corrective actions like training of staff) restrictions on further recruitment. In case of protocol violation and lack of reporting of study progress, the IEC asked for an explanation, with a clear warning not to repeat the violation in future and ensured that corrective actions were implemented. Additional AE reports were asked for from investigators who had failed to submit them in time. For the SAEs not reported earlier, follow-up reports and outcome were called for from the PIs and reviewed. Continued GCP training of all investigators was made compulsory and PIs were asked to submit a copy of the certificate at the time of submission of a new project. Moreover, in certain studies where it was felt that patients’ safety and well-being were compromised, IECs asked for monitoring reports from the sponsors and the recommendations of the data safety monitoring board, if relevant.
At sites with inadequate study supervision, the IEC advised recruitment of additional GCP trained members in the study team. It was recommended that at least one study coordinator per trial be appointed. In many investigator-initiated studies, resident doctors were the PIs, so the IEC recommended the training of resident doctors in clinical research. PIs who did not keep their documents securely under lock and key were asked to make arrangements to do so.
| Table 2: Action taken by IEC against monitoring sites. | |
| Violation theme | Action taken by IEC |
| Protocol deviation | Explanation asked for with a clear warning against future repetition |
| Deviation from investigational plan | Restriction on future recruitment,submission of audit reports from sponsor |
| Non-reporting ofstudy progress to IEC | Explanation asked for with a clear warning against future repetition |
| Deficiencies in study supervision | Recruitment of additional members in the study team advised |
| SAE reporting | Submission of AE reports |
| Lack of investigator awareness (protocol and ICD) | Continued GCP training of study recommended |
Discussion
The site monitoring visits carried out by the IECs of a tertiary care hospital in India revealed innumerable protocol violations, which would not otherwise have been identified. The findings relating to violations of the informed consent process were particularly disturbing as they violated the basic principle of autonomy, a fact that needs to be viewed seriously. There were discrepancies between the consent forms approved by the IEC and the forms used at the site; in some cases, ICDs, or the signatures of patients and /or PIs were missing. These violations were similar to those observed in the studies carried out by McCusker et al (8), in which an audit of 188 consent forms of 33 protocols revealed that consent forms were missing from the site, non-approved consent forms had been used, and the signatures of participants, witnesses and investigator were found missing in many forms. Another study by Smith et al (9) in 1997 showed that, of the 39 projects reviewed; a quarter had protocol deviations in relation to the consent process. The same study had found that, though adverse events had been reported, projects which were abandoned or late to start were vastly underreported to the IEC (9). In our study, delayed reporting of serious adverse events was a common finding.
Qualitative interviews with investigators for 16 research projects conducted by Douglass et al (10) concluded that an active monitoring programme can detect deviations from the approved protocol not disclosed in the annual report. The same was observed in our study during the interviews with the PIs.
An encouraging finding in our study was that the patients’ understanding of the research study, including the benefits and risks involved, was adequate. It was therefore felt that most of the protocol violations, including those related to the ICD, were due to an overload of clinical work rather than the unethical behaviour of the PI.
The results of this study reveal that there is an urgent need for an active monitoring programme by IECs for the continuing review of ongoing projects. Currently, lack of infrastructure, manpower, funds and time is a major hurdle for conducting active site monitoring. Most IECs spend a substantial amount of time in reviewing and approving protocols and reserve some time for passive monitoring, ie, reviewing SAE reports, periodic status reports, etc. There is very little time left to carry out site monitoring. Another aspect is the lack of training. IEC members are not trained to conduct monitoring.
In India, there is no central body which offers accreditation, trains IEC members to monitor studies, and monitors IECs for their compliance. In the studies by Demets and Weijer (5, 11) the authors have discussed the problems involved in carrying out continuing review by IECs. Smith et al (9) have reported that each detailed review takes six person hours at a cost of 120.
The need for recognition and affiliation of IRBs is currently not mandatory in India and depends, not only on the enthusiasm and motivation of an IEC, but also on the funds that it receives. Many IECs in India do not receive sufficient funding and the institutes in which they are situated are not keen to provide funding on a priority basis. IECs therefore continue to struggle to meet the responsibility of accreditation, continued training, and staffing.
Active on-site monitoring helped our IEC to identify problems related to the implementation of GCP which could not have been detected by the passive ongoing review of study-related documents carried out routinely by our IECs. Thus IECs need to have mechanisms for site monitoring in place so as to ensure that GCP is followed in letter as well as spirit.
Declaration: No competing interests nor funding from any external agency to be declared.
References
- Indian Council of Medical Research. Ethical guidelines for biomedical research in human participants [Internet]. New Delhi:ICMR;2006 [cited 2011 Nov 9].p.111. Available from: http://www.icmr.nic.in/ethical_guidelines.pdf
- Ministry of Health and Family Welfare, Department of Health. Drugs and Cosmetics (II amendment) rules, 2005 [Internet]. New Delhi: Government of India; 2005 Jan 20 [cited 2011 Nov 23]. Available from: http://dbtbiosafety.nic.in/act/Schedule_Y.pdf
- World Health Organisation. Operational guidelines for ethics committees that review biomedical research [Internet]. Geneva:WHO;2000 [cited 2011 Mar 26]. Available from: www.who.int/tdr/publications/publications/
- Pickworth E. Should local research ethics committees monitor research they have approved? J Med Ethics. 2000 Oct 26; 26(5):330-3.
- DeMets DL, Fost N, Powers M. An institutional review board dilemma: responsible for safety monitoring but not in control. Clin Trials. 2006;3(2):142-8.
- Morse MA, Califf RM, Sugarman J. Monitoring and ensuring safety during clinical research. JAMA. 2001 Mar 7;285(9):1201-5.
- Ethics committee for research on human subjects. Seth GS Medical College and KEM Hospital, Mumbai. Site monitoring visit [Intranet] 2009 Jun 26 [cited 2011 Nov 11]. Available from: www.kem.edu/dept/ethicscommittee/SOP15.PDF
- McCusker J, Kruszewski Z, Lacey B, Schiff B. Monitoring clinical research: report of one hospital’s experience. CMAJ. 2001 May 1;164(9):1321-5.
- Smith T, Moore EJH, Tunstall-Pedoe H. Review by a local medical research ethics committee of the conduct of approved research projects, by examination of patients’ case notes, consent forms, and research records and by interview. BMJ. 1997 May 31;314:1588-90.
- Douglass AJ, Jarvis A, Bloore S. Monitoring of health research by research ethics committees. N Z Med J. 1998 Mar 13;111(1061):79-81.
- Weijer C, Shapiro S, Fuks A, Glass KC, Skrutkowska M. Monitoring clinical research: an obligation unfulfilled. CMAJ. 1995;152(12):1973-80