THEME: CHALLENGES OF INDIA’S DRUG REGULATORY SYSTEM
RESEARCH ARTICLE
The quality challenge for generic medicines in India: An industrial policy-sensitive perspective
Dinesh Kumar Abrol, Rollins John, Nidhi Singh
Published online first on May 25, 2026. DOI:10.20529/IJME.2026.017Abstract
This article provides an industrial policy-sensitive understanding of the problem of quality of Indian generic medicines supplied both to the domestic market and to weakly regulated markets in Asia, Africa, and South America. Most of these medicines come from micro, small and medium enterprises (MSME). While all drugs manufactured in the country must comply with standards under the revised Schedule M of the Drugs and Cosmetics Act, 1945, the deadline for MSMEs’ compliance has been extended repeatedly, and even as of March 2026, drugs are manufactured in the country in two categories of manufacturing facilities — one compliant with the revised standards and another non-compliant with the revised standards.
While double standards are unacceptable, the policy discourse on medicine quality focuses entirely on uniformity of standards, and their regulation, without setting an industrial policy-sensitive context for the reasons for poor quality, and for developing an appropriate response.
We argue that the problem of medicine quality is closely connected to the structural changes in the industry after India signed the Agreement on Trade Related Aspects of Intellectual Property Rights. Large-scale enterprises depend on MSMEs — many of which are poorly equipped — for supplying branded generic medicines in the domestic market and poorly regulated markets abroad. Further, a sharp decline in the indigenous manufacture of raw materials and active pharmaceutical ingredients has left the industry vulnerable, because of its dependence on China for the import of these materials.
We propose that poor quality must be addressed through industrial policy-specific changes, institutional collaboration, and technical support, not merely by closing down MSMEs. Quality assurance cannot depend on a system of inspection alone; quality by design must be built into the manufacturing process, and there must be strict enforcement of standards.
Keywords: Industrial policy, generic medicine, quality by design, Indian pharmaceutical industry, Central Drugs Standard Control Organization, US Food and Drug Administration
Introduction
India has often been described as the pharmacy of the world. The Indian pharmaceutical industry supplies the bulk of generic drugs (meaning off-patent drugs, whether branded or unbranded) to countries across the world. Therefore, problems of medicine quality are of great concern both to India and to the other countries to which it supplies medicines.
In recent years, there have been many reports of poor-quality generic drugs from Indian manufacturers causing serious injury and death. Most of these reports are from the domestic market and from weakly regulated foreign markets of Asia, Africa, and South America; the bulk of medicines supplied here come from micro, small and medium enterprises or MSMEs, classified based on their investment size (up to ₹50 crore) and turnover (up to ₹250 crore).
In the case of deaths in the Gambia and Uzbekistan caused by adulterated cough syrups manufactured by Indian firms, investigations by the World Health Organization (WHO) found that the syrups contained unacceptable levels of diethylene glycol and ethylene glycol — both toxic substances [1]. The companies supplying these substandard medicines to those markets are known to be supplying similar products within India, as India has a similar epidemiological profile; this raises serious questions about the safety of medicines used by Indians as well. Indeed, this has been confirmed in the recent child deaths in Madhya Pradesh and Rajasthan, as well as earlier deaths of children in Kashmir, both following ingestion of adulterated cough syrup [2].
Ultimately, in most of these cases, the manufacturers were let off the hook or faced inadequate regulatory consequences, largely because the national regulatory authorities in these importing countries were perceived to be weaker, under-resourced, or lacking the technical capacity to ensure the quality of the drugs imported [3].
In India, too, regulatory responses to reports of adulterated medicines remain slow and fragmented, and accountability measures for the manufacturers are either delayed or insufficient.
Take the latest case of contamination of certain batches of “Coldrif” cough syrup which was linked to child deaths in Madhya Pradesh and Rajasthan. Even as the Union Health Ministry said that the cough syrup samples did not contain toxic contaminants, Tamil Nadu’s Drugs Control Department undertook tests and confirmed the presence of a toxic industrial solvent in the samples tested [4].
Prompt action by the Tamil Nadu government, the failure of the Union Health Ministry, and the dynamics of public deliberations reveal the complex challenges of industrial policy making and of building a sound regulatory framework in the context of federal governance. The strict ban on this batch of cough syrup, announced by the Punjab government and some other states also tells us that the strengths and potential of the federal regulatory framework can be proactively utilised through the collaboration of Central and State level regulatory agencies [5, 6].
At one level, the problem has been viewed as a question of generics versus branded medicines. That is to say, a brand name can be taken as a marker of the medicine’s quality. This was the view held by the late Bibek Debroy, Chairman of the Economic Advisory Council to the Prime Minister. He saw the problem of spurious and sub-standard medicines in the market as “a generic problem” — a problem of promotion of unbranded generic medicines — and wrote that the push for “unbranded drugs” was a bad idea. Forcing doctors to prescribe only unbranded generics, and selling only unbranded generics at Jan Aushadi outlets, because they were less costly, was unacceptable, he wrote [7].
However, brands per se cannot be relied upon to guarantee the quality of medicine. For one, most Indian companies employ MSMEs to manufacture branded off-patent medicines for the domestic market and for weakly regulated foreign markets.
The problem of quality of generic medicine is also viewed as a governance failure, originating in weak regulation, inadequate staffing of the regulatory system, and lack of coordination between the Centre and states. Calls for urgent collective action through better Centre-State coordination on the industrial licensing side, and enforcement of uniform good manufacturing practice (GMP), have been renewed in the Indian pharmaceutical industry [8]. Large pharmaceutical units of foreign and domestic origin are also participants in this call. Since state governments are empowered by the Indian constitution to issue industrial licences to pharmaceutical units, these large pharmaceutical units are promoting the narrative of “ease of business” to argue for constitutional change, and seeking a shift in the loci of industrial licensing and factory regulation from the state level to a centralised system [9].
In essence, this discourse on drug quality focuses on regulation without an industrial policy-sensitive framing of the reasons for poor medicine quality, and the appropriate corrective measures.
In this paper, we discuss drug quality in the context of changes in the Indian pharmaceutical industry following India’s signing of the Agreement on Trade Related Aspects of Intellectual Property Rights (TRIPS), and the 2002 Drug Policy. We also look at the challenges facing the industry today, in respect of improving the quality of generic medicines for the domestic market and for the poorly regulated markets in other parts of the world.
Methods
The study uses a public policy analysis based on public documents, government policy documents, and data provided by ministries and regulatory bodies. Primary data sources include Not Standard Quality (NSQ) classifications reported by the Central Drugs Standard Control Organisation (CDSCO) between 2022 and 2025, and inspection observations from the United States Food and Drug Administration (USFDA) databases for Indian firms during the same period. Secondary data include government policy papers, manufacturing guidelines, and government and WHO surveys of the industry. Previous research by the authors provides additional context for the analysis.
India’s drug policies and the industry’s shift from independence to contract manufacture
It is not well known that, during the 2000s, India officially dreamt of becoming a knowledge superpower, a major producer of innovator medicines, as well as an exporter — the pharmacy of the world — of generic medicine. The 2002 drug policy was introduced for this purpose. This was contrary to the 1978 drug policy which had focused on developing a self-sufficient domestic industry of affordable medicines prioritising the domestic market.
India’s 1978 drug policy [10] had defined key decisions of industrial policy for generic medicines, with the objective of reducing reliance on imported bulk drugs; encouraging indigenous production of raw materials and active pharmaceutical ingredients (APIs); and regulating the prices of drugs in the country. It reserved major areas in the domestic pharmaceutical market for either the public or the domestic private sector. It introduced drug price regulation to ensure the affordability of essential medicines. It stipulated that companies maintain in-house production capabilities for both bulk drugs and formulations, ensuring an integrated approach to pharmaceutical manufacturing. Finally, it encouraged firms to develop technological expertise across the entire production chain — from raw materials to finished drugs — thus strengthening the domestic industry’s capacity and also laying the foundation for India’s emergence as a global leader in the production of affordable generic medicines.
All this changed with the 2002 drug policy [11], which was introduced with the objective of obtaining an increase in the export footprint of Indian pharma in the United States (US) and European Union (EU) markets. Among the major changes instituted were: removal of all licensing requirements for certain formulations; abolition of licenses for some bulk drugs; permission to the domestic private sector to choose the directions in which it would invest; permitting 100% Foreign Direct Investment (FDI), and incentivising Outward FDI (investment in foreign enterprises with the objective of maximising pharma exports).
Large domestic pharmaceutical firms were offered extensive tax rebates, tax holidays, and financial support from the Council for Scientific and Industrial Research (CSIR) for product innovation. The 2002 policy had only incentives without penalties for non-compliance. These decisions resulted in a structural transformation of the industry.
Strategic choices of large pharmaceutical firms and erosion of domestic capabilities
Rather than opting for the path of structural competitiveness via strengthening technological and economic self-reliance, large firms chose to focus on outward foreign direct investment, much of which went down the route of highly import-dependent, low value-added generic medicine exports to regulated markets. In-house production of pharmaceuticals was limited to the regulated markets in the US and the EU.
The once largely self-reliant Indian pharmaceutical industry gradually became dependent on imported raw materials and APIs, particularly from China, to remain cost-competitive in the export of generic medicines to highly regulated markets such as the US and the EU.
As shown in the Supplementary Table, imports of bulk drugs and drug intermediates from China consistently accounted for more than 57% of India’s total bulk drug imports from FY14 onwards, rising to over 74% by FY25. This growing import dependence contributed to the progressive shrinkage of domestic API manufacturing capacity.
In parallel, large pharmaceutical firms increasingly outsourced manufacturing for the domestic market to MSMEs, reinforcing a fragmented production structure [12]. Major companies such as Pfizer and Dr. Reddy’s Laboratories contracted out the manufacture of several formulations — particularly price-sensitive generic products — to smaller firms located in industrial clusters [12: Table 6]. While this strategy helped large firms reduce costs and focus on regulatory-intensive export markets, it further weakened incentives for in-house API production and technological upgrading within the domestic pharmaceutical ecosystem.
In many cases, the large firms reduced their investment in manufacturing medicines for the domestic market and the weakly regulated markets abroad. They started using, on a regular basis, the provisions for loan licensing, contract manufacturing, and third-party manufacturing available in the Drugs & Cosmetics Act of 1945 [12]. These practices were transactional, poorly regulated, and exploitative of MSMEs. Many MSMEs followed the large firms and shifted their production to “pharma parks” or designated zones for MSMEs that were supposed to provide technical support for testing, as well as infrastructure such as warehouses, industrial effluent discharge facilities, incentivised by the tax exemptions given to these pharma parks. However, these parks were poorly equipped. Neither the pharma parks nor the pharma clusters have the required support from publicly funded facilities which can offer affordable services to MSMEs for testing, research, and consulting for operational excellence and quality improvements [13].
R&D and innovation crowded out by export-oriented expansion
The investment in research and development and product innovation expected to follow the 2002 policy did not materialise [12]. In the case of new medicines, India chose to encourage domestic firms to purchase voluntary licences from foreign firms rather than have the government issue compulsory licenses; only one compulsory licence has been issued in the last 23 years [14]. “Innovative medicines” are imported and far fewer are locally manufactured. India is becoming dependent on imports for the availability of newer medicines.
Instead of innovation, large firms have preferred to invest in brand building which guarantees them a price premium. They have prioritised the promotion of “off-patent generics” as branded medicines, and are known to spend a lot of money on doctors working in both public and private sectors to ensure that they prescribe their respective brands — many of which are irrational combinations — to make more money in a poorly regulated market.
MSME proliferation and the challenge of policy support
Contrary to the expectations of standard economic theory, the liberalisation of trade and investment and intellectual property in India did not lead to industrial consolidation; instead, the number of small businesses in the pharmaceutical sector increased in a big way.
While estimates of the number of pharmaceutical units vary, a government survey in 2023 records that the Indian pharmaceutical industry had 7,673 industrial units in 118 pharma clusters (with an average of 65 units per cluster) spread over 19 states/UTs [15]. Almost 88% of these units are MSMEs ― 1,995 micro industries, 2,393 small industries, and 2,331 medium scale industries. Only 954 of these units are large industries.
MSMEs form an essential part of the supply chain for the large industries and are the backbone of the pharma sector. They operate in the local market, and mainly manufacture and market formulations based on less complex molecules. Only 22% of MSMEs surveyed have availed of any subsidy or assistance under government schemes to support technical upgradation, manufacturing, or export [15].
Large firms extract greater profits from small firms through loan licensing and contract manufacturing [12]. A move by the government to scrap loan licensing was met with stiff resistance from industry associations [16], who argued that this would lead to revenue loss and closure of existing MSMEs, and medicine shortages.
A 2017 survey by WHO of 288 units manufacturing drugs and APIs, and 10 units engaged in the pharmaceutical trade [17], highlighted the following trends:
• Large firms were increasingly using contract manufacturers to serve the domestic market.
• Only about 44% units were producing their own brands/products; the remaining were involved in contract manufacturing, fulltime or part-time.
• Three out of five contract manufacturers interviewed reported difficulties in conducting their business. They worked with narrow profit margins and faced delays in payments.
• Small firms were also trying to survive using contract manufacturing while looking for tie-ups with large firms to stay viable.
• Only 120 of the 288 units had their own drug testing lab facility.
• Contract manufacturers reported difficulties because of delays in payments by large firms, and low profit margins that could compromise quality.
• Firms mentioned delays in supply of finished products, and questions of quality assurance.
Among the WHO report’s recommendations are the need for standardisation and re-examination of contract manufacturing processes to ensure more uniform and transparent procedures [17].
Shifts in the manufacture of medicines: how and for whom
Pharmaceutical quality is closely linked to sustained investments in APIs, quality control testing, compliance with Good Manufacturing Practices (GMP), skilled manpower, and process validation. When the contract manufacturer is forced to keep production costs very low to make a profit, firms may respond by sourcing cheaper inputs, reducing testing frequency, underinvesting in upgrades, or stretching equipment utilisation, all of which can compromise product quality.
As seen in Table 1a, the share of the total cost of production (COP) of drug formulations to serve the domestic market (as opposed to the export market) fell from nearly 70% to below 39% between 1997 and 2016. The share of cost of production associated with exports of drug formulations and services rose.
Table 1a. Changing patterns of Cost of Production*, 1997-2016
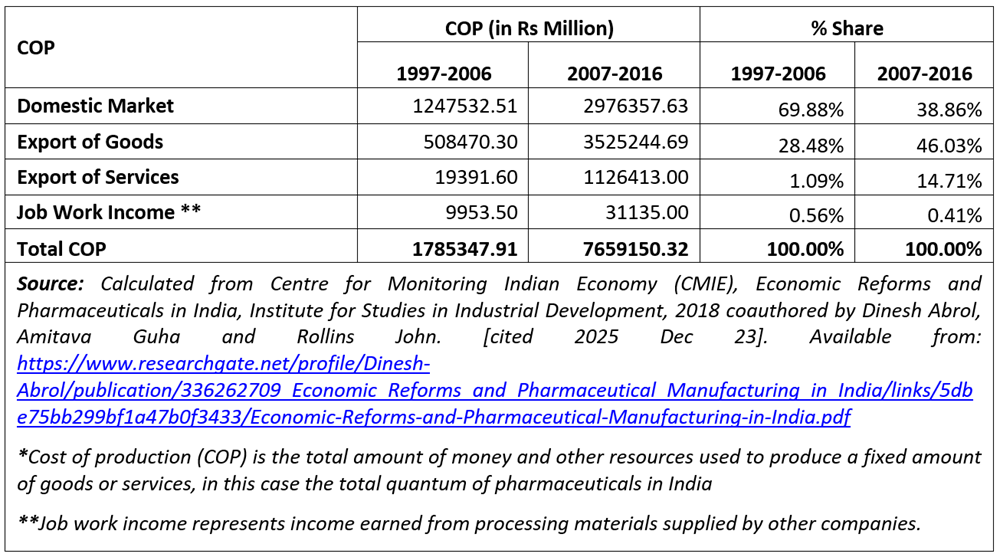
Table 1b gives the changing patterns of cost of production and sale of goods, disaggregated by market segments, for the periods 1997–2006 and 2007–2016.
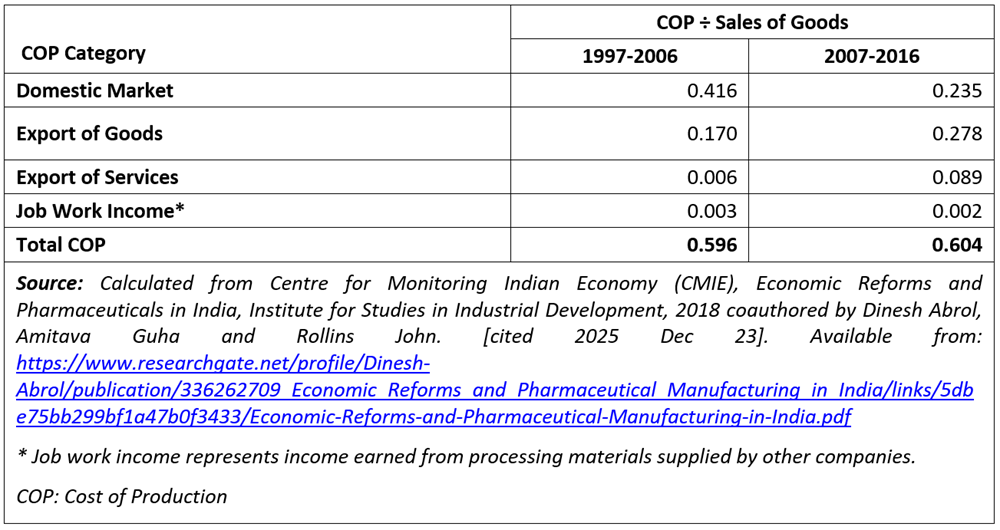
While the ratio of aggregate production cost to sales remains broadly stable at around 60% across both periods, we observe that manufacturers reallocated production costs across different markets. In the case of sales in the domestic market, the ratio of production cost/sales fell from 0.416 in the first period to 0.235 to the second period; but in the case of the export markets the ratio rose from 0.170 to 0.278 over the same period. These contrasting trends are revealing. Such a drive to reduce production costs for medicines in the domestic market — because companies are not investing in fixed costs, or they are buying cheaper raw materials, or they are employing unskilled workers at lower wages — is bound to affect the quality of those medicines.
From a quality-of-medicines perspective, ensuring investment for GMP compliance and securing investment from large as well as small enterprises in local manufacturing are important challenges for industrial policy.
Insufficient investment in manufacturing for the domestic market, and dependence on imported raw materials without complete testing, are contributing to inconsistent drug quality and posing public health risks. As companies relocate to remote parts of the country which offer tax incentives there are concerns about whether these units possess the infrastructure, facilities, and workforce to manufacture quality medicines compared to units in large industrial regions. There has also been a relaxation in oversight requirements, along with shortages of staff and inadequate infrastructure.
Moreover, under different loan licensing agreements, drugs can be manufactured according to different standards in the same facility. Thus, it is possible for the same unit to make drugs for the US and EU following GMP, while also making drugs for other, poorly regulated export markets, and for India, without the same standards of quality control [3].
It has also been highlighted that India has a large domestic market for fixed drug combinations (FDCs) — many of which are irrational mixes of existing drugs. The promotion of branded generics for product differentiation purposes has encouraged irrational drugs and poorer quality drugs. This has become a preferred means of sustaining profitability for both large companies marketing well-established branded medicines and MSMEs who have functioned below the radar of regulatory oversight in general. FDCs are useful only when they enhance therapeutic efficacy, lower side effects, offer pharmacokinetic benefits, improve compliance by reducing pill burden, minimise doses, prevent resistance, or lower costs. Poorly designed FDCs can be harmful [18].
Quality and regulatory issues
As per the Drugs & Cosmetics Act, 1940, a drug is labelled “Not of Standard Quality” or poor quality if it fails to meet the standards prescribed in the relevant section of the Act, or in one of the official pharmacopoeias. A drug can be labelled NSQ because it is misbranded, spurious, or adulterated. Unlike in well-regulated markets, where drug quality is evaluated by visits to factories, examining the documentation of the manufacturing process as well as testing of samples before they enter the market, enforcement of quality in India largely takes place at the marketplace where the regulator takes medicine samples off the pharmacy shelf and tests them.
Our analysis shown below confirms that poor-quality drugs continue to be a significant problem for both the domestic and export markets.
Table 2 with data from CDSCO for 2022 to 2025, shows that the number of Indian firms notified for Not Standard Quality (NSQ) reports went from 4 in 2022 to 183 in 2025. The reasons for an NSQ report were categorised under misbranding, non-compliance with laboratory protocols, and non-compliance with protocols specified in the Indian Pharmacopeia. These 567 firms will include large manufacturers, small scale manufacturers, and merchant exporters (traders who buy and sell medicines without participating in the manufacturing process).
Table 2. Trends in non-standard quality reports from CDSCO (2022-2025)
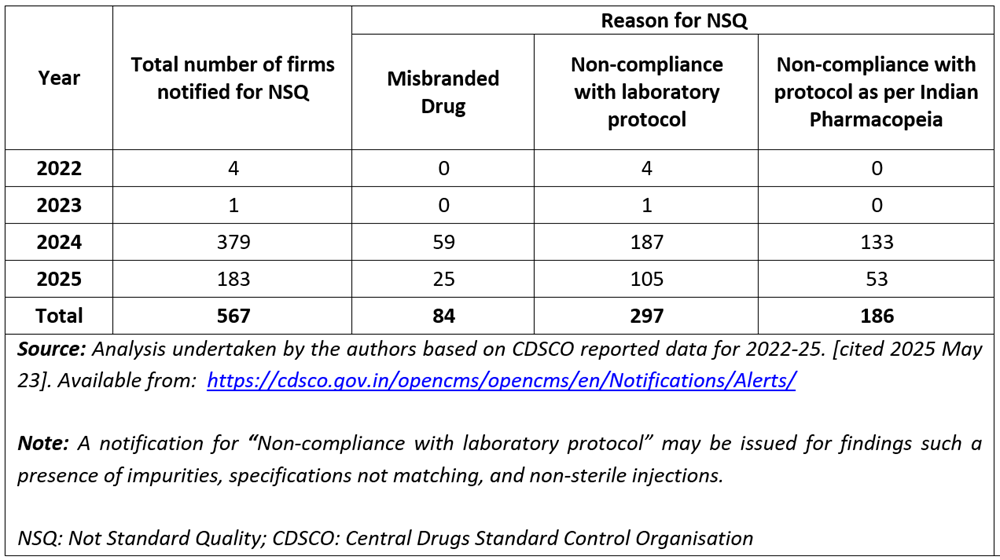
Table 3a on Indian firms notified by the USFDA for NSQ cases 2022-2025 reveals patterns in regulatory compliance and quality management issues. The number of firms under scrutiny rose from 47 in 2022 to 138 in 2023, peaking at 159 in 2024 before declining to 55 in 2025. This trend may reflect a combination of intensified regulatory scrutiny during the mid-years and potentially improved compliance, or fewer inspections in 2025.
Table 3a. Trends in classification of violations by USFDA (2022-2025)
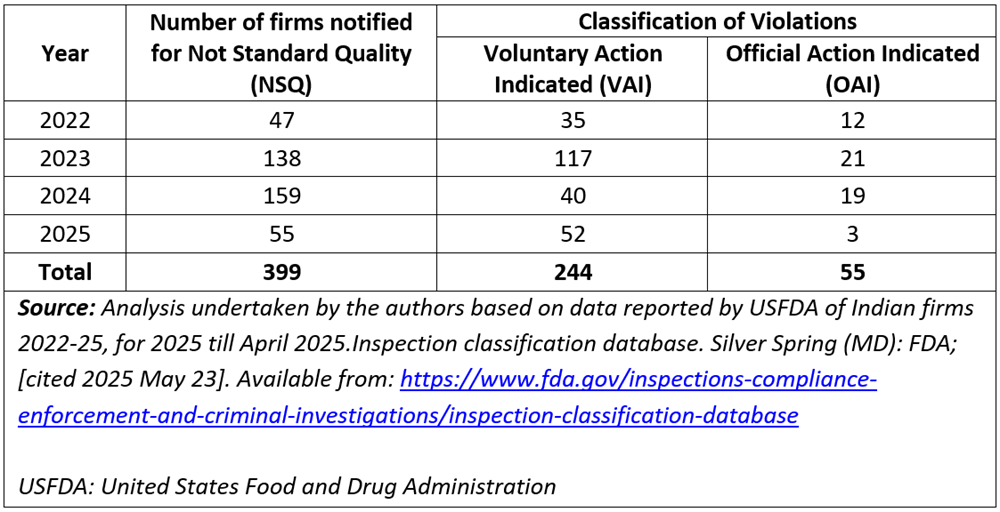
The USFDA classifies violations as Voluntary Action Indicated (VAI) or Official Action Indicated (OAI). The OAI designation is for serious lapses that could result in formal regulatory actions such as warning letters or import alerts. A substantial proportion of these 399 (244/399) cases fell under the VAI category, where firms were expected to take corrective action without facing immediate enforcement. In 2022, 35 out of 47 firms were classified as VAI, and in 2025, 52 out of 55 fell into the same category — suggesting that most firms could address observed deficiencies.
Table 3b on the nature of the deficiencies provides further insights. Lack of documentation consistently appeared as a concern across the years. Other recurring issues included the absence of a clear responsibility structure, inadequate quality control units or procedures, and, in some cases, poor manufacturing infrastructure. These point to systemic weaknesses in internal governance and operational processes. A significant number of NSQ observations — particularly in 2023 and 2024 — were marked under “reason not given” (88 and 110 cases, respectively), suggesting either limited disclosure or complex violations that defied easy categorisation.
Table 3b. Nature of deficiencies in non-standard quality (NSQ) reports from the USFDA to Indian Firms 2022-2025
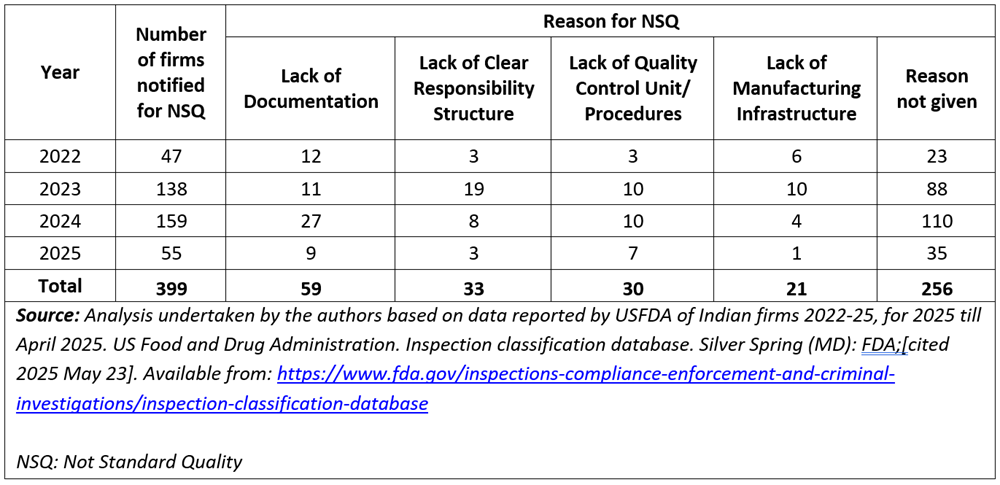
Beyond the numbers, what stands out is the nature, depth, and categorisation of quality issues identified by the USFDA compared to the CDSCO, suggesting a deeper, systemic issue within India’s domestic drug regulatory framework.
The USFDA’s observations span a wide range of foundational deficiencies — from lack of documentation and unclear responsibility structures to inadequate quality control procedures and insufficient manufacturing infrastructure. These are not minor lapses but core failures in pharmaceutical manufacturing and regulatory adherence. The USFDA’s classification of violations as requiring either voluntary action or official action offers a transparent and graduated response structure that encourages both accountability and corrective action. In contrast, CDSCO’s reporting appears narrower in scope and less rigorous. Most CDSCO NSQs are categorised as misbranding or non-compliance with laboratory protocols (eg, impurity presence, non-sterility, or mismatched specifications).
Structural and procedural lapses — such as absence of documentation or poor internal quality systems — are largely missing from CDSCO’s classification, suggesting either they are not routinely inspected for, or not consistently documented.
This discrepancy between the CDSCO and USFDA findings points to an underlying regulatory capacity gap in India. Multiple studies and expert analyses have highlighted that the Indian drug regulatory system is often understaffed, undertrained, and under-resourced relative to its global counterparts [3,19]. In contrast to the Indian regulatory system, stringent regulatory authorities such as the USFDA employ risk-based inspections, in-depth audit trails, and electronic documentation reviews, allowing them to detect issues proactively and systematically.
The USFDA prioritises inspections based on the potential risk to public health posed by a facility, product, or process. Facilities or products considered higher risk (for example, sterile injectables, biologics, or drugs with a history of quality issues) are inspected more frequently or thoroughly. While the Indian pharmaceutical industry has made significant strides globally, especially as a supplier of generic medicines, domestic regulatory oversight is not keeping pace with international expectations. This poses reputational and public health risks, particularly as Indian firms expand their presence in regulated markets, besides being a major ethical concern.
What quality control entails
Unlike the focus on quality by inspection at the marketplace currently followed by Indian regulators, the framework used by regulatory authorities in well-regulated markets is that of “quality by design”. This ensures medicine quality by integrating it into the design of the manufacturing process. Qualified human power, knowledge of the product and the manufacturing process, materials from approved vendors, production and marketing according to the conditions of the license, and following other applicable regulations are all essential ingredients for quality by design. Also built in is a continual improvement in quality. A regular review of quality, a “root cause analysis” of defective products, periodic management reviews, and a quality manual should be in place, and documentation of these steps should be available for scrutiny by drug inspectors. An integral feature of this process is quality risk management, which demands documentation of review of all batches that failed — investigations and corrective and preventive actions taken, complaints and recalls, the qualification status of equipment, etc.
Good manufacturing practices or GMP ensure that products are consistently produced according to the quality standards appropriate to their intended use and as required by the license. GMP are incorporated in the revised Schedule M which includes new elements reflecting a quality by design approach, such as requirements for premises, utilities and equipment and processes; analysis of faulty manufacture and product deterioration; reporting of serious quality problems, changes in facilities and processing steps; tests required for checking quality of materials and storage systems, the role and responsibility of the licensor, the details of the agreement, documentation of evidence of technology transfer between the licensor and licensee, etc. All of this is to be made available in writing to the authority issuing the license to manufacture the drug.
The revised Schedule M also states that the manufacturer “must assume responsibility for the quality of the pharmaceutical products”. It also speaks of the need for a “pharmaceutical quality system” covering suppliers and distributors, and requires “management of outsourced activities.” It mandates that “arrangements are made for the manufacture, supply and use of the correct starting and packaging materials, the selection and monitoring of suppliers and for verifying that each delivery is the correct material from the approved supply chain.” The revised Schedule M also explicitly mentions “outsourced activities” and requires that control and oversight be part of the quality management system.
What is needed in proper quality assurance
In the framework of quality by design, the challenge of testing raw materials, and a systemic response, came up in the recent case of cough syrups involving diethylene glycol, with important lessons for framing industrial policy.
Industrially, glycerol IP (glycerine) is usually produced with vegetable oils, using a chemical process known as saponification. It can also be synthesised from organic compounds using feedstock from the hydrocarbon industry. Glycerol is also a waste product from processes used to manufacture biodiesel from fats. The nature of impurities arising from these different processes is different. As some processes generate impurities like ethylene glycol and diethylene glycol, the raw material testing protocol needs to be stringent.
There are some 40 manufacturers of glycerol IP in India, but its availability in the retail market has been compromised, at least since November 2018.
A case was described by Amit Misra, retired scientist from Central Drug Research Institute (CDRI), Lucknow (2021, correspondence available with DA). M/s Neurochem, an MSME, tested a sample of glycerine IP. It passed the required test as a borderline case but the company asked for further testing with the Common Research and Technology Development Hub at CDRI. The sample passed the test for purity, but showed unexpected signals in the infra-red spectrum. Further analysis by gas chromatography and mass spectroscopy found significant amounts of ethylene glycol and diethylene glycol present. The results of the tests were shared among MSMEs by word of mouth, and potential tragedies were avoided.
This instance reveals the potential contribution of publicly funded research institutions and their collaboration with pharmaceutical MSMEs. The availability of technical support can be an incentive for MSMEs to improve their products’ quality. MSMEs’ decisions should be built into the system (quality by design). Thus, the establishment of cluster-based research and technology hubs as essential common facilities must be a mandatory requirement by both Union and State governments for industrial policy framing.
Industrial policy and its future directions
The government is currently trying to get industry to prioritise implementation of revised Schedule M guidelines [20].
Even though the transition period has ended, compliance levels among MSMEs remain low. It is estimated that nearly 5,000 units face closure if no further reprieve is granted. There are ongoing discussions between industry bodies and the government for a potential further one-year extension.
However, implementation of guidelines alone is not likely to work, as the current state of pharmaceutical quality is an illegitimate second order impact of the 2002 industrial policy changes. Similarly, the current relationship between MSMEs and large firms is a product of the response of domestic pharmaceutical firms to the 2002 policy. Large and small firms in the pharmaceutical industry coexist, both as competitors as well as cooperating entities. It is not the existence of MSMEs but the relationship between small and large firms that is a problem and needs to be changed. Complete success in this regard will require changes in industrial policy to get the industry to prioritise operational excellence wherein quality and safety, rationality and affordability would be the utmost concerns.
Joint liability
The first step is to act on the current double standard system. MSMEs should not be permitted to further postpone the implementation of the revised Schedule M. At the same time, manufacturers must assume responsibility for the quality of the medicines they market. The administrative decision of making large firms and MSMEs jointly liable was taken as far back as 2020, and when the product is made in an MSME and marketed by a large firm, the marketer along with the manufacturer is held responsible for medicine quality [21, 22]. The strict enforcement of joint liability will push large and small firms to collaborate and work together to produce quality medicines for the domestic market.
Institutional support
The CDSCO envisions the revised Schedule M as a strategic tool to align domestic pharmaceutical manufacturing practices with global standards, particularly those set by the WHO. While large pharmaceutical firms have either already met, or are better equipped to meet, these enhanced standards — often due to prior investments aimed at satisfying stringent regulatory authorities like the USFDA or EMA — many smaller firms face significant hurdles. Lacking both financial and technical resources, many small-scale manufacturers are struggling to stay afloat, despite government support schemes. This disparity highlights the urgent need for stronger institutional support and targeted interventions to ensure that smaller players are not left behind in the push for global quality compliance.
Currently there is an underusage of the Department of Scientific and Industrial Research-established platform of common research and technology development hubs for assuring quality of raw materials. Dependence on imports of key raw materials is going to continue if the government persists with a policy of permitting private investment without requiring the industry to develop and strengthen local manufacturing capacity. CDSCO and the law leave this challenge to MSMEs which do not have the wherewithal to assure quality of imported or locally produced raw materials. Centralised testing of raw materials can support MSMEs to ensure quality and meet standards.
Cluster-level support
In parallel, cluster-level credit facilitation cells, jointly supported by the Department of Pharmaceuticals, state governments, and the Small Industries Development Bank of India, could provide financial and technical handholding for smaller enterprises undertaking technology and quality upgrades. To support MSMEs’ transition to the requirements of the Revised Schedule M guidelines, the Pharmaceutical Technology Upgradation Assistance Scheme has also now been revamped, resulting in the approval of 103 applications for financial assistance totalling ₹105 crore. The scheme offers varying levels of reimbursement based on company revenue, incentivising MSMEs to upgrade their facilities.
Restructuring cluster development schemes
Pharmaceutical clusters have benefitted from union and state governments under the cluster development programme with infrastructure and common facilities such as effluent treatment plants and testing centres. These should be used to set the industry’s house in order in respect of pharmaceutical quality and production arrangements. Pharma unit clusters developed around chemistry-sensitive public research support and common facilities can facilitate collaboration between large and small units. The licensing authority must ensure that MSMEs shift to clusters supported by pharmaceutical park facilities, with R&D support, if they wish to remain in business. At present, not all units are in clusters, and not all clusters are supported by pharmaceutical parks.
Regulation
The policy of handing out incentives and tax concessions for cluster development without regulation is not going to work. MSMEs will have to upgrade their facilities and adopt globally recognised GMP without perceiving regulatory reforms as punitive. Quality-linked soft loans and interest subvention schemes — tied to demonstrable progress in meeting revised Schedule M requirements or obtaining WHO-GMP certification — would help distribute the cost of compliance more equitably.
It is also necessary to institutionalise collaboration between the National Institutes of Pharmaceutical Education and Research and the Pharmacy Council of India for the development of a certified regulatory workforce programme that would provide pre-service and in-service training for regulatory inspectors, quality assurance officers, and compliance managers; set national competency standards in GMP, quality risk management, and pharmacovigilance; and ensure periodic re-certification.
Training
We need to strengthen the process of training MSMEs and ensure that neither MSMEs nor large-scale firms cut corners. Only qualified human power may be recruited to operate manufacturing facilities. It is also important to disincentivise large firms from poaching of technical personnel of MSMEs, a practice that is preventing large and small firms from sincerely collaborating for industrial upgrading.
Quality assurance
Such interventions would lead to robust quality assurance systems within Indian pharmaceutical manufacturing. They also show how the industrial research system needs to be engaged to generate advisories and alerts to ensure the attention of bulk manufacturers towards following additional precautions/processing steps/downstream processing steps required to ensure that the product meets the Indian Pharmacopoeia standards. Quality standards must be universally upheld regardless of the market in which the product is sold. Without meaningful oversight and consequences for violations, vulnerable populations in less-regulated markets continue to bear disproportionate risks, raising serious ethical and public health concerns.
The effectiveness of this initiative is hindered by challenges such as limited coordination between the CDSCO and State Drug Regulatory Authorities, as well as chronic understaffing across regulatory bodies. The drug regulatory system must be better staffed and equipped.
India’s pharmacovigilance framework, though institutionally established, remains underutilised — contributing less than 2% of global Adverse Drug Reaction reports to the WHO’s VigiBase, despite being one of the world’s largest producers and exporters of pharmaceuticals. Future iterations should integrate a discussion on pharmacovigilance as an essential component of the regulatory ecosystem.
These are not small steps. The industry will have to adjust to these changes, and the drug regulatory system will have to function and maintain its integrity. MSME closure is not a solution. The Indian state and society nurtured the domestic industry, MSMEs formed the domestic industry, and large Indian firms have emerged from among the MSMEs. The industrial policy will have to reemphasise an integrated local production system, and the priority must be to produce essential and rational drugs. The drug policy of 1978 was embedded in the vision and strategy of integrated production. With suitable modifications, the vision and strategy of 1978 should be reworked for the policy on pharmaceutical manufacturing in 2026.
Recommendations
To sum up, we propose the following steps for implementation to address the crisis of quality in the Indian pharmaceutical industry:
First, the timeline of the end of December 2025 announced for the compliance of the revised Schedule M by the MSMEs should be strictly adhered to by the government. The revised Schedule M ensures the implementation of a quality by design philosophy and the related standards to be followed in manufacturing.
Second, the strict enforcement of the newly announced provision for joint liability between the unit that manufactures the drug and the company that markets it is necessary to ensure that even large-scale companies are accountable and held responsible for the compliance of the revised Schedule M by the MSMEs, when they depend on MSMEs to supply medicines for market development.
Third, the industrial policy framework needs to be revisited to encourage domestic manufacturing of critical raw materials and intermediates/inputs for bulk drugs to develop certified vendors of raw materials and bulk drugs.
Fourth, the cluster development scheme should be utilised to provide infrastructure for collaboration of large firms and MSMEs to provide required technical support to MSMEs and operationalise manufacturing excellence at enterprise level in cases where large scale companies are marketing medicines manufactured in MSME premises.
Finally, these steps must involve all those affected by the current situation. Following the recent reports of deaths from contaminated drugs, a committee was set up with the involvement of various industry bodies. However, there is no representation from MSMEs. The changes in the post-economic reform industrial structure, and the challenges of realignment of regulatory oversight, need more inputs both from small enterprises and civil society organisations.
Endnote: Loan licensing and its variantsIn loan licensing, a company with a licence but without a manufacturing facility can obtain permission to have its products manufactured in a facility owned by another manufacturer. In contract manufacturing, a brand-owning company enters into a legal agreement with another firm to produce, test, pack and label drugs under its brand name. The contract manufacturer is responsible for compliance with good manufacturing practices while the brand-owning company handles marketing and distribution. In third-party manufacturing, multiple companies may get their products made by a single manufacturer. The manufacturer may produce drugs for several brands simultaneously. This system is widely used in India’s pharmaceutical industry to reduce costs and expand production capacity. These provisions under the Drugs and Cosmetics Act of 1945 enable flexibility and scalability in drug production but also require strong regulatory oversight to ensure product quality, uniformity, and compliance with safety standards.
Authors: Dinesh Kumar Abrol (corresponding author — dinesh.abrol@gmail.com), Faculty, Transdisciplinary Research Cluster on Sustainability Studies (TRCSS), Jawaharlal Nehru University, New Delhi, INDIA; Rollins John (rollinskjohn@gmail.com), Member of Research Team, Transdisciplinary Research Cluster on Sustainability Studies (TRCSS), Jawaharlal Nehru University, New Delhi, INDIA; Nidhi Singh (nidhisinghb8@gmail.com), Assistant Professor, Parul University, Vadodara, Gujarat, INDIA.
Conflict of Interest: None declared Funding: None
To cite: Abrol DK, John R, Singh N. The quality challenge for generic medicines in India: An industrial policy-sensitive perspective. Indian J Med Ethics. 2026 Apr-Jun; 11(2) NS: 98-107. DOI: 10.20529/IJME.2026.017
Submission received: June 12, 2024
Submission accepted: August 20, 2025
Published online first: March 25, 2026
Theme Editors: Sandhya Srinivasan, Veena Johari
Peer Reviewer: An anonymous reviewer
Copyright and license
©Indian Journal of Medical Ethics 2026: Open Access and Distributed under the Creative Commons license (CC BY-NC-ND 4.0), which permits only noncommercial and non-modified sharing in any medium, provided the original author(s) and source are credited.
References
- BBC. Gambia child deaths: WHO stands by ‘dangerous’ India cough syrup claim. 2022 Dec 16 [Cited 2025 Dec 8]. Available from: https://www.bbc.com/news/world-asia-india-63996180
- Mollan C. WHO flags regulation gaps after India child deaths from cough syrups. BBC.com. 2025 Oct 10 [Cited 2025 Dec 5]. Available from: https://www.bbc.com/news/articles/c3vzgz794wro
- Krishnan V, John A. The massive failures of India’s drug regulatory system. Pulitzer Center. 2025 Mar 12 [Cited 2025 Dec 20]. Available from: https://pulitzercenter.org/stories/massive-failures-indias-drug-regulatory-system
- ET Online Bureau. Regulator finds toxic contaminant in cough syrup linked with deaths in MP. Economic Times. 2025 Oct 4 [Cited 2025 Dec 20]. Available from: https://pharma.economictimes.indiatimes.com/news/policy-and-regulations/regulator-finds-toxic-contaminant-in-cough-syrup-linked-with-deaths-in-mp/124309010
- Bhirani G. Punjab bans Coldrif cough syrup following deaths of children in Madhya Pradesh. 2025 Oct 7[Cited 2025 Dec 20]. Available from: https://www.livemint.com/news/india/punjab-bans-coldrif-cough-syrup-following-deaths-of-children-in-madhya-pradesh-11759822875921.html
- HT correspondent. Punjab bans ‘adulterated’ Coldrif syrup after 16 deaths in Madhya Pradesh. Hindustan Times. 2025 Oct 7[Cited 2025 Dec 20]. Available from: https://www.hindustantimes.com/cities/chandigarh-news/punjab-bans-adulterated-coldrif-syrup-after-16-deaths-in-madhya-pradesh-101759820151442.html
- Debroy B. Generic drugs everywhere: that is a problem. Indian Express. 2023 Oct 12 [Cited 2025 Dec 12]. Available from: https://indianexpress.com/article/opinion/columns/bibek-debroy-generic-drugs-everywhere-problem-8978921/
- Bukhari A. Toxic Cough Syrup, Weak Oversight: India’s Unending Drug Safety Crisis. Drug Policy Watch. 2025 Oct 15 [Cited 2025 Dec 20]. Available from: https://healthpolicy-watch.news/toxic-cough-syrup-weak-oversight-indias-unending-drug-safety-crisis/#:~:text=Share%20this:,such%20disasters%20to%20keep%20unfolding
- Chandna H. Cough Syrup Tragedy Calls for Regulatory Reform: Wockhardt Chief Habil Khorakiwala Tells News18. News18. 2025 Oct 8[ Cited 2025 Dec 22]. Available from: https://www.news18.com/india/cough-syrup-tragedy-calls-for-regulatory-reform-wockhardt-chief-habil-khorakiwala-tells-news18-ws-l-9622108.html
- Statement on Drug Policy of 1978 placed before the Lok Sabha by the then Union Minister of Petroleum, Chemicals and Fertilisers, Shri HN Bahuguna. 1978 Mar 29[Cited 2025 Dec 20]. Available from: https://drive.google.com/file/d/1k360wzWWtzBMfuZU8155_-GXUootePIh/view?usp=sharing
- National Pharmaceutical Pricing Authority, Union Ministry of Chemicals and Fertilisers. Pharmaceutical Policy 2002. 2002 Feb 15[Cited 2025 Dec 20]. Available from: https://nppa.gov.in/uploads/pdf/PHARAMACEUTICAL-POLICY-2002pdf-4045093fbf76f4142a1f33dc2d859337.pdf
- Abrol DK, Rollins J, Guha A. Economic Reforms and Pharmaceutical Manufacturing in India. 2018. New Delhi: Institute for Studies in Industrial Development. ISBN 978-81-938075-2-1
- Das S. Deadly Silence: India’s Negligent Pharma Manufacturing Units. BioSpectrum India. 2025 Jul 31 [Cited 2025 Dec 10]. Available from: https://www.biospectrumindia.com/features/73/26437/deadly-silence-indias-negligent-pharma-manufacturing-units.html
- Saha PK, Mukherjee A. Compulsory Licensing of Pharmaceutical Patents in India A Policy Shift. Econ Polit Wkly. 2019 Feb 2[Cited 2025 Dec 20]. Available from: https://www.epw.in/journal/2019/5/commentary/compulsory-licensing-pharmaceutical.html
- Department of Pharmaceuticals, Union Ministry of Chemicals and Fertilisers. Survey of pharma clusters. New Delhi: Centre for Market Research & Social Development; 2023[Cited 2025 Dec 12]. Available from: https://pharma-dept.gov.in/sites/default/files/Final%20Report-Survey%20of%20Pharma%20Clusters.pdf
- Trivedi I. Govt Proposal to Scrap Loan Licensing in Draft Pharma Policy Faces Flak. LiveMint. 2017 Sep 11[Cited 2025 Dec 20]. Available from: https://www.livemint.com/Industry/q8sAJkHB6v3tfdi0akImHO/Govt-proposal-to-scrap-loan-licensing-in-draft-pharma-policy.html
- World Health Organization. Country Office for India. Survey of Indian pharmaceutical enterprises for meeting national and global health needs. New Delhi: WHO; 2017[Cited 2025 Dec 22]. Available from: https://cdn.who.int/media/docs/default-source/searo/intellectual-property/survey-of-india2016 Jul-Aug;n-pharmaceutical-enterprises-for-meeting-national-and-global-health-needs—nov-2017.pdf?sfvrsn=5cbe96f5_1
- Gupta YK, Ramachandran SS. Fixed dose drug combinations: Issues and challenges in India. Indian J Pharmacol. 2016 Jul-Aug;48(4): 347-349. https://doi.org/10.4103/0253-7613.186200
- Hotkar Singh India’s drug safety deficit: critical gaps in an overly complex system need urgent remedies. The Hindu. 2025 Aug 26 [Cited 2025 Dec 23]. Available from: https://www.thehindu.com/sci-tech/health/indias-drug-safety-deficit-critical-gaps-in-an-overly-complex-system-need-urgent-remedies/article69974974.ece
- Press Information Bureau, Ministry of Health and Family Welfare, Govt of India. Conditional extension of timeline to small and medium pharmaceutical manufacturers for compliance with revised Schedule M notification. New Delhi: PIB; 2025 Feb 12[Cited 2025 Dec 12]. Available from: https://www.pib.gov.in/PressReleaseIframePage.aspx?PRID=2102291
- Babu G. Health ministry to amend D&C Rules to issue licenses to marketers to ensure quality & safety of drugs. Pharmabiz. 2025 Nov 29[Cited 2025 Dec 12]. Available from: https://www.pharmabiz.com/NewsDetails.aspx?aid=182729&sid=1#:~:text=+%20Font%20Resize%20–,Health%20ministry%20to%20amend%
- Ministry of Health and Family Welfare, Govt of India. Gazette of India. GSR 101(e). 2020 Feb 11 [Cited 2025 Dec 12]. Available from: https://cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadGazette_NotificationsFiles/gsr101E)_Definition,%20Responsibility%20s.pdf