ARTICLE
Reports of site monitoring visits by institutional ethics committees in an Indian tertiary care hospital: A retrospective analysis
Yashashri C Shetty, Kritarth Naman M Singh, Padmaja A Marathe, Sharmila V Jalgaonkar, Snehalata Gajbhiye, Janhavi Katkar, Manali U Vengurlekar
DOI: 10.20529/IJME.2019.042Abstract
The monitoring of clinical trials is an integral function of the institutional ethics committee (IEC)to ensure the ethical conduct of research. The National Ethical Guidelines for Biomedical and Health Research Involving Human Participants, 2017, of the Indian Council of Medical Research, underline a strong need for active monitoring of clinical trials. A previous study by the authors, of research studies initiated between 2008 and 2010, had found many lapses after site monitoring. In the present study, 12 clinical studies—both sponsored and investigator initiated—were monitored by members of the King Edward Memorial Hospital (Mumbai) IEC between 2011 and 2017. The most common violations seen were related to informed consent (8/12 sites). The other violation themes were lack of investigator understanding of protocol (6/12), deviation from the investigational plan (5/12), non-reporting of the study’s progress to the IEC (4/12), and patient recruitment prior to IEC approval (2/12). The IEC took various corrective actions, such as ordering retaking of consent and good clinical practice (GCP) re-training and requiring interim reports, explanations for deviations, upgradation of facilities, and payment of pending compensation. The IEC even froze review of protocols from a frequently defaulting Principal Investigator’s (PI) site and put study recruitment on hold for the same PI. This study demonstrates that active site monitoring by IECs is a must for ensuring the ethical conduct of studies.Introduction
Monitoring, the act of overseeing the progress of a clinical trial, helps to ensure that the trial is conducted, recorded, and reported in accordance with the trial protocol, standard operating procedures (SOPs), good clinical practice (GCP) guidelines, and the applicable regulatory requirements (1). The Declaration of Helsinki mentions that ethics committee must have the right to monitor ongoing studies so that they can be conducted in the best way possible (2). In the United States of America (US)ethics committees are entrusted with the initial review of proposed interventional research protocols prior to project initiation and with the continuing responsibility of regular monitoring for the necessary ethics compliance until study completion (3).
Until recently, it was not compulsory for ethics committees in India to actively monitor clinical studies. However, it is heartening to see that the situation is changing. The office of the Drug Controller General of India has monitored institutional ethics committees (IECs)by site inspection and by reviewing the re-registration forms of ethics committees with the Central Drugs Standard Control Organisation (CDSCO), which considers monitoring of investigator sites as a pre-requisite to ensuring the ethical conduct of clinical studies. Numerous updates to policy and guidelines governing clinical research in India have been introduced by the Indian regulatory authorities, namely the CDSCO, the National Accreditation Board for Hospitals and Healthcare Providers (NABH), and the Quality Council of India for Ethics Committees (4). Subsection 1.4.5 of the NABH accreditation standards stipulates that monitoring of trials be done to ensure equitable selection of participants, with special attention to vulnerable and high-risk participants (4). However, there are no parameters to ascertain whether IECs are able to achieve their objective of patient protection in clinical research.
IECs perform passive monitoring by reviewing the review reports, protocol violation reports, safety reports, and completion reports submitted to them by the study team at regular intervals. However, this form of “passive monitoring” may not reflect the real-life scenario of a clinical trial (3),(5). Though passive monitoring is done regularly by IECs, according to the 2017Indian Council of Medical Research (ICMR)guidelines, there is a great need for “active monitoring” as well (6).
Site monitoring procedures have existed in the IEC SOP of the King Edward Memorial (KEM) Hospital, Mumbai since November 14, 2008. There are two ethics committees functioning in KEM Hospital (7). The authors have previously performed a study to record the IEC monitoring practices at KEM Hospital from 2008 to 2010and found several lapses at the sites (5). The present study is a follow-up to the previous study and was conducted to analyse the reports of site monitoring visits made by the IEC over the past seven years (2011–2017), and compare the more recent practices with past findings (2008–2010). This paper discusses the consequent recommendations made to the concerned investigators as also action taken by the IEC against the study teams.
Methodology
Twelve clinical studies, both sponsored and investigator‒initiated, were monitored by the members of the KEM IEC over a period of seven years (2011–2017). The monitoring was conducted using a standardised format, in accordance with the SOPs of the KEM IEC. The monitoring was routine as well as for cause. Site visits were conducted according to the IEC SOP number 12 (7). The principal investigators (PIs) were informed in writing two weeks prior to the scheduled site visits. PI availability as well as acceptance were confirmed before conducting the site visits.
The visiting team consisted of two IEC members, who noted down their observations in their report Review of documents was conducted in the IEC office. The site monitoring forms for the projects—including the initial letters with monitoring findings sent by the IEC to the investigators, along with the responses of the investigators—were reviewed. Later, the follow-up letters sent by the IEC were also reviewed. The identities of the investigators, sponsors, and monitors of the studies were not noted, and confidentiality was maintained by all the members of the site monitoring teams.
The reports were analysed for violations and categorised under the following themes:
1. Informed consent issues
2. Deviation from investigational plan<
3. Non-reporting of study progress to IEC
4. Deficiencies in study supervision by investigator
5. IEC approval status
6. Lack of investigator’s understanding about protocol and informed consent document (ICD)
7. Serious adverse event reporting
8. Other findings: No source documents found; no coded drugs used; documents not kept under lock and key; auditors’ monitoring report missing, biodata of investigators in the project file not signed
The findings of the present study were analysed by descriptive statistics and compared with the results of the 2008-2010 study conducted by Shetty et al (5).
Results
Violation themes
Out of the twelve studies monitored in this study, seven were sponsored by actors within the pharmaceutical industry, while five were investigator‒initiated clinical studies. The most common violation seen was related to informed consent (8 of 12 sites; see Table 1 for full findings).
| Table 1. | |||
| Sr. No. | Violation Themes | Monitoring Sites | |
| 1 | Informed consent issues | 8 | |
| 2 | Lack of investigator’s understanding about protocol and informed consent document | 6 | |
| 3 | Deviation from investigational plan | 5 | |
| 4 | Non-reporting of study progress to IEC | 4 | |
| 5 | Patient recruitment before IEC approval to IEC | 2 | |
| 6 | Other findings: • No source documents found • Documents not kept under lock and key • PI reported SAE late • Compensation for SAE not paid by sponsor • PI was not eligible to continue in the study as superannuated and not a permanent employee of the institute |
5 These issues were noted one per site |
|
IEC=institutional ethics committee; PI =Principal investigator; SAE=Serious adverse events
Informed consent process
At eight of the twelve sites violations were found, related to the informed consent process. A wrong version of the informed consent document (ICD) was used at one site, not approved by the IEC. At one site, language errors were present in the ICD, with overwriting noted as well. Another issue related to the informed consent process was the presence of legally acceptable representatives’(LARs) signatures in two ICDs, along with the signatures of the study participants.When a participant is mentally sound and conscious, there is no need to take a LAR’s signature. In some ICDs, the names of the participants or the investigators were found missing. At one of the sites, the approval date of the translated version of the ICD was not found.
A participant at one trial site was interviewed to assess participants’ understanding of the study. This site had previously been advised by the IEC to re-obtain consent based on an earlier consent violation. It was found that the participant had a good understanding of the trial after the repeated consent procedure.
Lack of investigator understanding about protocol
At six of the twelve study sites, the IEC monitoring teams found that there was a lack of understanding about the study protocol and/or the ICD. On questioning the principal investigator (PI) and the team about the inclusion criteria, they were not able to state the contents of inclusion criteria. While reviewing their consenting process it was found that AV consent had been taken of a patient who was HIV positive. This is prohibited under the Schedule Y regulations, which stipulate that audio consent of HIV positive patients has to be taken to maintain confidentiality. In this context, we found the investigators were unaware of the said guidelines regarding AV consenting.
Deviation from study plan
Five of the study teams had deviated from the original study plan. Most of these cases were related to lack of knowledge regarding the proper consenting process, as the monitoring team found ICD related issues in four of these studies. At two sites, the wrong version of the ICD was used for consent procedures, and hence the monitoring team found a need to properly retrain the study team in these cases. In the study plan, we mentioned that the approved version of the consent form would be used , but here a prior version of the ICD was used instead of the final version approved by the Ethics Committee, amounting to a protocol violation .One site did not carry out electrocardiogram (ECG) tests in three patients for whom cardiac outcome was an area of investigation, thereby leading to possible safety concerns. At the same site, the necessary changes had not been made in the source documents, which were previously ordered by the IEC. At one study site, the study team had deviated from the inclusion criteria for recruitment of patients, thereby leading to a major study violation.
Non-reporting of study progress to IEC
At four of the 12 study sites, the monitoring team found that the progress reports were not submitted by the study teams on time. Pharmaceutical studies are monitored at regular intervals and a monitoring report is submitted to the PI. These reports should be submitted to the Ethics committee for review. In this case, the study team members did not submit the monitoring report, and were unable to show the monitoring report during the site visit.
Patient recruitment before IEC approval
At one of the study sites, it was found that 10 participants out of the required 14 had been enrolled in the study before the IEC granted study approval. Similarly, at another study site, it was found that all the required 60 participants were recruited into the study before obtaining IEC approval. This shows that the study teams had a lack of understanding regarding the importance of IEC approval.
Other findings
Other issues reported by the IEC monitoring team at three different study sites included absence of source documents at the study site, improper storage of trial-related documents at the study site, late or no reporting of the SAE by the study team, and failure to pay compensation to patient(s) for SAE by the sponsor. The study site forgot to report SAE to the Ethics Committee and the participant took reimbursement from his personal insurance company. As he did not have the original bills, the sponsor’s insurance company did not reimburse him the admission charges. The sponsor and the investigator were not fulfilling their roles. In one of the studies, the principal investigator (PI) was no longer a permanent employee of the institution as PI annulment had already transpired. However, the study had been ongoing and the IEC had not been informed about this development. There was no PI oversight in that study.
Comparison of violation themes observed during site monitoring in 2011–2017 with the previous study of 2008–2010
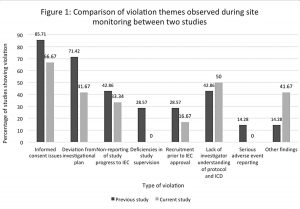
The most common violation theme in both the present study (2011–2017) and the previous study (2008–2010) was found to be related to informed consent. There was an increase in the percentage of cases that had issues with investigator understanding of the protocol and the ICD in the present study (50%) as compared to the previous study (42.86%); (see Fig 1 for the full details). None of the study sites were found to have deficiencies in study supervision in the current monitoring, as compared to the monitoring done previously, where such deficiencies were found (28.57%). There was a surge in some new violations in the current monitoring (41.67%), as compared to the previous monitoring report, such as not informing the IEC about PI annulment, and recruiting more participants than the number approved by the IEC. The other findings in the current audit monitoring included lack of source documents at the study site, improper storage of the study documents, late reporting of SAE, and failure to give compensation to patients who suffered SAE or adverse events (AE).The AEs are supposed to be managed by the PI and the sponsor; but here the PI did not manage the AEs nor report them to the Ethics Committee. Figure 1 depicts the incidence of various violation themes in both the new and the previous monitoring.
Action taken by the IEC
Subsequent to data collection, the site monitoring teams prepared and presented their reports at the next IEC meeting. The findings of the monitoring teams were sent in writing to the PIs of the respective studies, with an instruction to establish compliance within one month. The PIs established compliance and submitted their reports to the IEC. In these reports, the PIs explained how the violations affected participant safety and what steps they have taken to prevent protocol deviations. After reviewing these compliance reports, the IEC gave approvals and recommendations to the PIs to continue with the studies.
In studies where the monitoring team found issues with the informed consenting process or the ICD, reobtaining consent from participants was recommended by the IEC. It was also suggested that fresh consent documents be submitted to the IEC. At study sites where protocol awareness was the identified issue, the whole team was asked to undergo re-training in protocol as well as GCP. The study teams were asked to explain protocol deviations, and to submit timely interim reports in cases where the interim reports were earlier delayed. At the two sites where recruitment of participants was done prior to obtaining IEC approval, use of the patient data sets was disallowed and a complete repetition of the enrolment process was ordered. The PIs were instructed to make the necessary changes in the source document, wherever needed. Another recommendation was for the facilities at the study sites to be upgraded for proper maintenance of study documents. One of the PIs—who was found responsible for late reporting of SAE and non-payment of compensation when related to the study—was directed to pay compensation to the patients. The PI was also required to furnish the IEC with proof of payment of compensation.
At one of the study sites, the PI was found to have been involved in five different lapses, in the examined study as well as in multiple earlier studies. This PI was not even a permanent employee of the institution at the time of the monitoring visit. The study was ongoing, but the IEC had not been informed about the PI’s retirement. Totally, twelve cases of protocol deviations were found in the same department, of which the IEC had not been informed, relating to SAE compensation not being paid, and to inclusion of more patients than the approved number. Hence, it was decided that no study protocol from that department would be reviewed by the IEC for the next three months; and the recruitment procedures for all ongoing studies in that department would also be suspended for three months.
The corrective actions taken in the present study and those taken in the 2008-2010 study by Shetty et al (5) are documented in Table 2.
Table 2: Corrective actions in present study (2011–2017) vs previous study (2008–2010)
| Table 2. | |||
| Corrective actions taken in present study | Corrective actions taken in Shetty et al (5) | ||
| • Reobtaining of participant consent ordered • Explanations required for deviations from approved study plan • Study team instructed to undergo GCP retraining • Interim reports required to be submitted on time • Repetition of participant recruitment ordered • Necessary changes in the source documents recommended • Facility upgrade for maintaining study documents recommended • Payment of compensation to patients reporting AE/SAE ordered, and proof of payment required • In one case, owing to multiple lapses by a PI and at the site, 3-month embargo placed on protocol review in that department as punishment, and recruitment for ongoing studies withheld | • Explanations required for violations and clear warning issued against any future violations • Audit reports and progress reports required to be submitted on time • Recruitment of additional members to the study team advised to remedy deficiencies in study supervision • AE reports required to be submitted on time • Continued GCP training recommended for the whole study team | ||
Discussion
Multiple protocol violations were reported by the monitoring teams in our study. This was similar to the findings of the 2008-2010 study by Shetty et al (5), which had monitored studies approved by the same two IECs of the same institution. It is important to note that issues related to the informed consent process, deviation from investigational plan, non-reporting of study progress to the IEC and recruitment prior to IEC approval were much fewer in the current study as compared to those found in the previous study by Shetty et al, thus indicating better trial execution between 2011 and 2017.
A study by Douglass et al (8) undertaken in New Zealand echoes the same finding and insists on active on-site monitoring to find deviations that cannot be identified through passive monitoring. Ochieng et al, in a study in Uganda, found lapses in informed consent documentation; for 25% of ongoing studies, annual updates were not submitted to their IECs (9). They recommend on-site compliance monitoring as possibly the most appropriate method to minimise such non-compliance, though often ignored by IECs, purportedly due to lack of capacity and high maintenance costs, both in terms of human and financial resources. Uganda has 14 accredited IECs that review and approve research, but only four have reported carrying out monitoring of approved studies at the site—and only one was found to have records at the site as evidence of conducting on-site compliance monitoring (9). Deficiencies in study supervision, which were found at 28.57% study sites in the 2008-2010 study by Shetty et al, were totally absent in our current study. This shows that there has been an improvement in the study management and regulation by the sponsors and the investigators over the past few years.
Some new issues—absence of source documents, improper and unsafe storage of documents, late reporting of SAEs, and non-payment of compensation for AEs—were identified by the IEC in this study while monitoring clinical trial sites that were not seen in the earlier study by Shetty et al. This shows the need for facilities at trial sites to properly store trial-related documents and to make the study team aware of the importance of reporting and compensating patients suffering from any AE. IECs must check site and investigator before issuing approvals so that such deviations are avoided.
There were a few positive findings noted by the site monitoring teams in our study. At one of the study sites, it was found that the study team had complied with the recommendations given by the monitoring team on a previous trial. This shows that monitoring of study sites does help in improvement of trial conduct, and hence it should be done in both passive and active form. At another study site, a participant was interviewed to ascertain his understanding of the study, and it was found that there was no coercion or lapse in the recruitment procedure in that study. This indicates that study teams may be ethical in their approach towards participant recruitment and trial conduct; but still commit technical ICD violations often. This could probably be due to clinical work overload, as these are public hospitals.
Despite the few positive findings, the plethora of protocol violations suggests an urgent need for an active monitoring programme by IECs to continue review of ongoing projects. IECs need to have mechanisms for site monitoring in place to ensure studies are being conducted incompliance with the protocol, SOPs, regulatory guidelines, and GCP. There are many hurdles to executing active site monitoring, including lack of infrastructure, workforce, funds, and time.
One potential solution is for IECs to have an internal monitoring board that monitors all funded and more-than-minimal-risk studies for which no external monitoring is mentioned in the protocol. IECs can also train their members to help them monitor clinical trial sites in a better and more efficient way. A study by Smith et al at Dundee, Scotland (10) had recommended that at least 10% of projects should undergo onsite review, with all others being monitored by questionnaire. They calculated that it required six person-hours of time and a salary bill of £120 per study monitored. They selected, at random, a stratified sample of 39 of the 311 projects approved in one year and found similar deviations as in our study.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use has, in its recent guidelines (11), emphasised risk-based monitoring for sponsors along with site monitoring. However, though the guidance documents mention site monitoring, the mechanisms to be adopted by IECs have not been detailed (11). Regulatory studies are monitored by sponsors through data safety monitoring boards or by regulators, but academic studies have to be monitored by IECs for oversight. IECs are overburdened because they have to monitor both academic and regulatory studies as our 2008-10 study depicts that the IEC was monitoring both academic and pharmaceutical driven studies (5). Pickworth, speaking from the perspectives of the US and Australia, where active monitoring is done by IECs, echoes some of our findings. Of the 39 projects approved by the Tayside Committee on Medical Research Ethics, they randomly monitor research projects by questionnaire and by site visits. Their monitoring reports published in 1997 showed that 9 studies of 39 had been discontinued but only one of these was reported as discontinued to the committee (3), (10).
In our present study, active trial site monitoring helped the IEC to identify violations. Many of these issues were impossible to identify by passive monitoring, underlining the importance of active monitoring. Though our study findings are similar to those of the previous study by Shetty et al (5), some new issues were noted in our current study, highlighting a need for regular active monitoring at study sites in future as well. We believe this will ensure patient safety and data credibility. Though the challenges in setting up an IEC and ensuring its smooth functioning make site monitoring seem like a herculean task, our example shows that it is not impossible.
A significant limitation of the current study is that it is a retrospective analysis of monitoring reports and corrective actions taken by a specific IEC. Therefore, these data cannot be generalised to other setups. As routine monitoring is not a norm for IECs, the sites selected for monitoring were primarily those with deviations reported.
Conclusions
The study found that active monitoring can bring out protocol deviations which are not reported. We believe that with proper training and financial investment, active site monitoring can be enhanced to risk-based monitoring for ultimate participant safety and protection. IECs should take institutional guidance and assistance to generate resources for this purpose, which can be the way forward.
Declaration regarding prior publication of similar work
A study of ethics committee monitoring practices at the King Edward Memorial Hospital, Mumbai, between 2008 and 2010, was published in the Indian Journal of Medical Ethics [Shetty YC, Marathe P, Kamat S, Thatte U. Continuing oversight through site monitoring: Experiences of an institutional ethics committee in an Indian tertiary-care hospital. Indian J Med Ethics. 2012 Jan-Mar;9(1):22-6]. The present study is a follow-up to that previous study, aiming to analyse the reports of site monitoring visits made by the Institutional Ethics Committee over the past seven years (2011–2017) and to compare current practices with past findings (2008–2010).
Conflict of interest declaration
The authors have no competing interests or funding from any external agencies to declare. This study was initiated after receiving IEC exemption IEC(II)OUT/324/17, dated March 23, 2017 from IEC-II of KEM Hospital.
References
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Integrated addendum to ICHE6(R1): Guideline for good clinical practice E6(R2). Current Step 2 version dated 11 June 2015[cited 2018 Nov 10] . Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Addendum_Step2.pdf
- World Medical Association. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA. 2013 Nov 27; 310(20):2191-4.
- Pickworth E. Should local research ethics committees monitor research they have approved? J Med Ethics. 2000 Oct;26(5):330-3.
- National Accreditation Board for Hospitals and Healthcare Providers (NABH) Accreditation standards for clinical trial in India ethics committee, investigator, and clinical trial site; 2015 [cited 2019 Jul 20] Available from: https://nabh.co/ClinicalTrial.aspx
- Shetty YC, Marathe P, Kamat S, Thatte U. Continuing oversight through site monitoring: experiences of an institutional ethics committee in an Indian tertiary-care hospital. Indian J Med Ethics. 2012 Jan-Mar [cited 2019 Nov 18];9(1):22-6.https://doi.org/10.20529/IJME.2012.006.
- Indian Council of Medical Research. National ethical guidelines for biomedical and health research involving human participants. 2017. [cited 2018 Nov 10]. Available from: https://www.icmr.nic.in/sites/default/files/guidelines/ICMR_Ethical_Guidelines_2017.pdf
- Institutional Ethics Committee, Seth GS Medical College and KEM Hospital. Site monitoring visit [SOP Document]. 2018 [cited 2018 Jul 25]. Available from http://www.kem.edu/wp-content/uploads/2018/07/SOP-12-Site-Monitoring-Visit.pdf
- Douglass AJ, Jarvis A, Bloore S. Monitoring of health research by ethics committees. N Z Med J. 1998 Mar 13;111(1061):79-81.
- Ochieng J, Ecuru J, Nakwagala F, Kutyabami P. Research site monitoring for compliance with ethics regulatory standards: review of experience from Uganda. BMC Med Ethics. 2013 Jun5; 14:23.
- Smith T, Moore EJ, Tunstall-Pedoe H. Review by a local medical research ethics committee of the conduct of approved research projects, by examination of patients’ case notes, consent forms, and research records and by interview. BMJ. 1997 May 31;314(7094): 1588-90.doi: 10.1136/bmj.314.7094.1588.
- ICH Harmonised Guideline: Integrated Addendum to ICH E6(R1): Guideline For Good Clinical Practice E6(R2) International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. 2015 Jun 11. [cited 2018 Nov 10. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Addendum_Step2.pdf