ARTICLE
Multiple ethical review in North–South collaborative research: the experience of the Ebola-Tx trial in Guinea
Maaike De Crop, Alexandre Delamou, Johan Van Griensven, Raffaella Ravinetto
DOI: https://doi.org/10.20529/IJME.2016.022 Published online: January 25, 2016Abstract
The process of double ethical review involves the ethics committees (ECs) in the country(ies) of the research site(s) and of the sponsor. This paper aims to assess the experience of the double ethical review in the Ebola-Tx trial, and to make general recommendations for research conducted during public health emergencies. The Ebola-Tx trial (ClinTrials.gov NCT02342171), sponsored by the Institute of Tropical Medicine in Belgium, was carried out at the Ebola Treatment Center of Médecins Sans Frontières in Donka, Guinea. The protocol was submitted to the national EC in Guinea, and to five more review boards of the sponsor and research partners. It took 55 days to get it approved. Some aspects were considered by at least three ECs, eg the informed consent and the ethical implications of the study design and exclusion criteria. Issues such as the fate of biological samples and capacity-building of local researchers were considered by one EC. The reviews complemented each other, thus raising the quality of the research and affording greater protection to the participants and community. In our experience, the double ethical review should be implemented routinely in externally sponsored trials. But in “urgency” situations, direct dialogue among the ECs should be fostered. Joint reviews would be greatly beneficial, but they would be feasible only if ad hoc mechanisms were planned before the emergence of a public health emergency.Background
Double ethical review
The “double ethical review” of externally-sponsored trials, ie ethical review carried out both in the country(ies) where a study is being carried out and in the country(ies) of the sponsor and research partners, has been widely proposed as a way of improving the protection of the participants and communities involved in the trial. For instance, it is recommended in the Ethical Guidelines for Biomedical Research Involving Human Subjects of the Council for International Organisations of Medical Sciences (CIOMS) (1) and in the report on The Ethics of Research Related to Healthcare in Developing Countries of the Nuffield Council on Bioethics (2). Double ethical review has also been explicitly requested by independent research groups (3, 4). Some regulators from sub-Saharan Africa took this line of thought further and considered the possibility of joint ethical reviews. For instance, the Guidelines of the Ethiopian National Research Ethics Review Committee (NRERC) (5) affirm that in collaborative research, “to avoid duplication of review efforts by IRBs, the NRERC may choose to conduct joint reviews in part or in whole, accept the review of another qualified IRB, or make other arrangements to establish oversight responsibilities”. The Uganda National Guidelines for Research Involving Humans as Research Participants affirm that in collaborative research projects, “each participating organisation is responsible for safeguarding rights and welfare of research participants. This involves securing research ethics committee approvals in both the local and foreign organisation. Where desirable, participating organisations in a collaborative research project may have a joint review arrangement for that particular research project” (6). Nonetheless, most national laws and regulations do not list double ethical review as a mandatory requirement in externally sponsored trials. Therefore, there is still a need to assess the double ethical review and provide evidence on its advantages and disadvantages.The double ethical review in the West Africa Ebola outbreak
Since March 2014, West Africa, in particular Guinea, Sierra Leone and Liberia, has been ravaged by the worst outbreak of Ebola Virus Disease (EVD) that has ever been witnessed (7). As of June 25, 2015, the number of laboratory-confirmed cases of Ebola recorded in Guinea was 3267, of whom 62.4% died (8). One of the key factors contributing to the high mortality rates was the lack of a proven effective EVD-specific treatment. Acknowledging the urgent need for clinical research to test candidate vaccines and therapies, in September 2014 the World Health Organisation (WHO) convened a consultation on vaccines and therapies that had demonstrated promising results in animal models for the prevention and treatment of EVD (9). The consultation resulted in a consensus that appropriate protocols should be rapidly developed for testing the most promising candidate interventions. It also stated that the investigation of innovative therapeutic approaches to EVD requires a concerted effort by multiple institutions based in different countries, and that “flexible approaches are required to harmonise various review processes, and ensure that the various ethics committees can review the projects simultaneously and share and discuss the review outcomes with each other”. To our knowledge, it is not yet known whether and how this specific recommendation has been implemented in the ethical review of externally sponsored clinical trials carried out for the current Ebola outbreak.The Ebola-Tx trial
Among the interventions identified during the September 2014 consultation, the assessment of the use of convalescent whole blood (CWB) and convalescent plasma (CP) was prioritised. This recommendation was driven by several reasons, including the pragmatic reality that – in contrast to most novel experimental drugs the supply of which was limited – the therapeutic value of CWB and CP could be evaluated within a short time span, and their widespread use for therapy could be implemented rapidly if proven effective. A pivotal document was drafted for use by national health authorities and blood transfusion services: “Use of Convalescent Whole Blood or Plasma Collected from Patients Recovered from EVD for Transfusion, as an Empirical Treatment during Outbreaks” (10). To help address this urgent research need, a research consortium was created to assess the efficacy and safety of CP in EVD cases. The consortium is led by the Institute of Tropical Medicine (ITM), Antwerp, Belgium and brings together 17 northern and southern institutions (Table 1). At the end of 2014, it obtained a grant from the EU Framework Programme for Research and Innovation Horizon 2020 to carry out the Ebola-Tx trial (Emergency Evaluation of Convalescent Plasma for Ebola Viral Disease in Guinea). This is an emergency, phase 2/3, open-label, non-randomised, clinical trial that evaluates the use of CP together with standardised supportive care (SC) in patients with confirmed EVD. Its primary objective is to assess if CP+SC improves the 14-day survival of patients, compared to SC alone. The ITM was the regulatory sponsor of the trial. The trial was carried out at the Ebola Treatment Centre (ETC) of Médecins Sans Frontières (MSF), Belgium in Conakry, Guinea, and it was registered in ClinTrials. gov as NCT02342171. Unlike other therapeutic trials, which evaluate the efficacy and safety of investigational medicinal products, the trial intervention (the plasma) in the Ebola-Tx comes from Ebola survivors in Guinea. Thus, this study involves two different vulnerable groups: the Ebola survivors who donated the plasma and the Ebola patients who received it.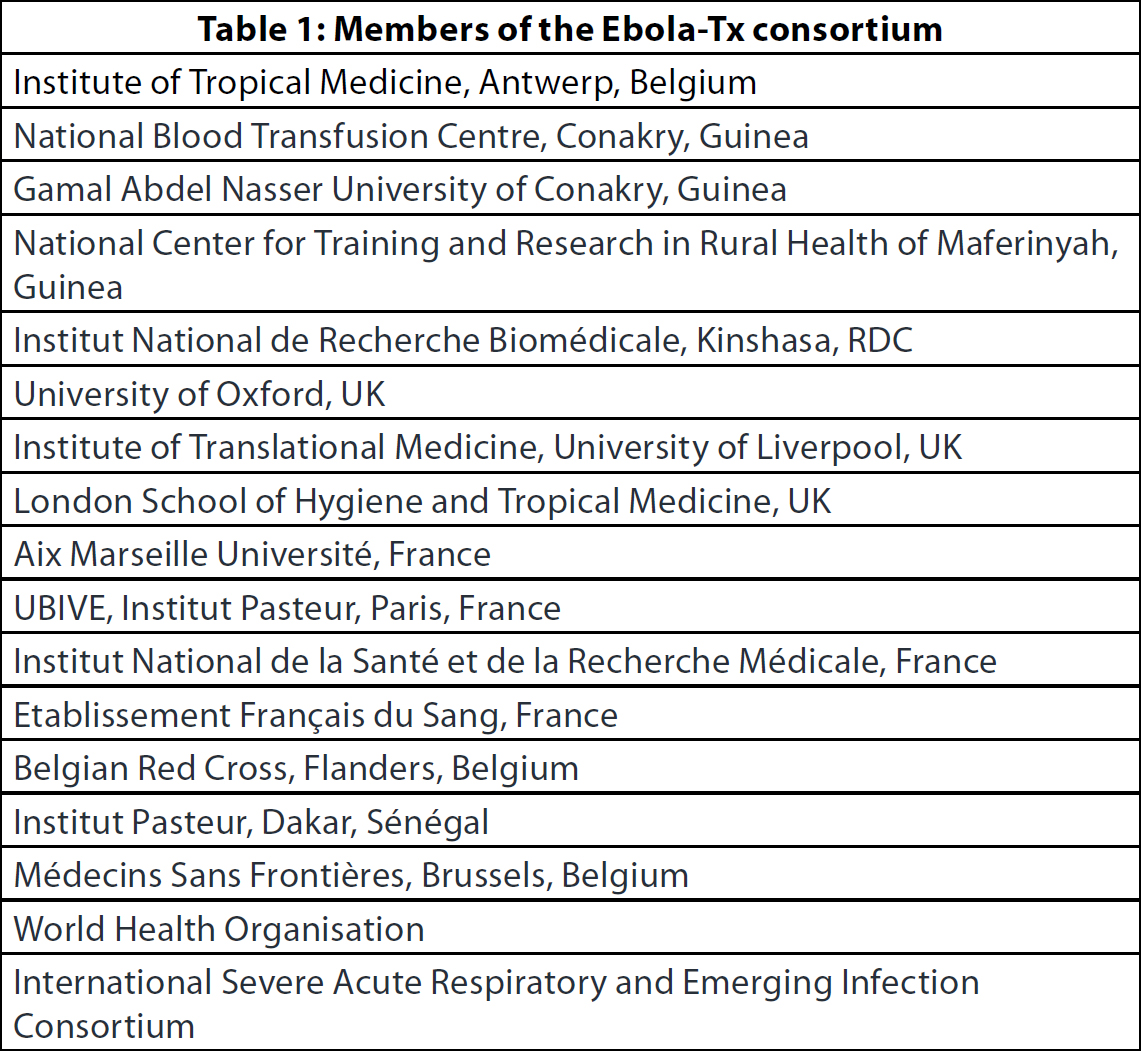

Objective
The primary objective of this paper is to assess the experience of the double ethical review in the Ebola-Tx trial, and to make general recommendations related to the usefulness and implementation of the double ethical review in externally sponsored trials. The secondary objective is to assess whether and how the recommendation from the WHO Background Document of September 2014 (“flexible approaches are required to harmonise various review processes, and ensure that the various ethics committees can review the projects simultaneously and share and discuss the review outcomes with each other”) has been implemented in the Ebola-Tx trial, and to make general recommendations for future outbreaks.Methodology
The process of the double ethical review in the Ebola-Tx trial was assessed by the trial coordinators in Belgium and Guinea, on the basis of an in-depth analysis of the documentation of the submission and review. In particular, we looked at:- the similarities and differences between submission requirements across the different ECs, as an indicator of the harmonisation or non-harmonisation of procedures in the double ethical review;
- the timelines for the reviews, as an indicator of the efficiency of the double ethical review process; and
- the contents of the reviews, to assess whether there were complementarities, contradictions or redundancies across the reviews of different ECs.
Findings
Submission requirements
The documents requested by the different ECs were quite similar. Each received copies of the protocol, the informed consent documents, the no-fault insurance certificate and the curricula vitae of the scientific coordinator and the country principal investigator. One EC also requested copies of the case report forms. Committee-specific questionnaires or templates were to be filled in for the majority of ECs (4/6). There were major differences across these templates, which ranged from simple checklists, to an on-line submission form including both checklists and narrative texts, to the template of the MSF ethics review board (ERB), based on a series of open-ended questions (11). As for the submission methods, one EC used a web-based submission platform, requiring log-in credentials; three required the submission package on paper as well as an electronic copy; and the two remaining ECs received all documents electronically only.Timelines for review
Table 3 gives an overview of the timelines for the submission and review process. First, in line with the sponsor’s internal procedure, the pre-final version of the protocol was sent to the IRB of the ITM. Given the urgency on account of the outbreak, the ITM IRB issued its comments immediately and discussed them in person with the study scientific coordinator, rather than waiting for the next scheduled meeting. This allowed for a very quick review (three days to approval). The protocol and informed consent documents were modified accordingly and the final version 1.0 was issued on December 2, 2014.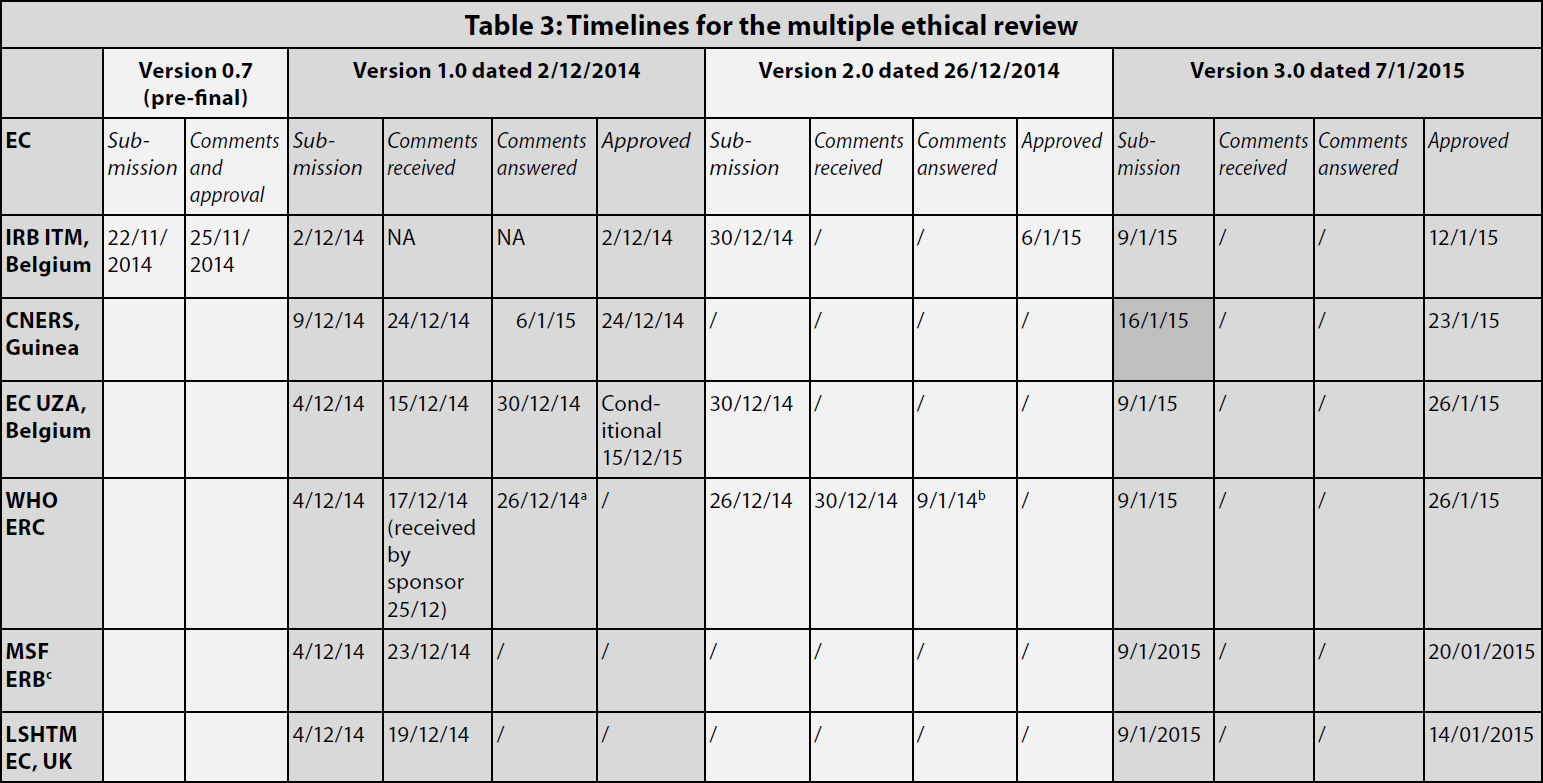
aThe comments to version 1 were addressed concomitantly to the submission of version 2.
bThe comments to version 2 were addressed concomitantly to the submission of version 3.
cThe given date is the date when the submission package was sent to the MSF research coordinator (who then did the submission).
Contents of review
Table 4 gives an overview of the main issues raised during the ethical review.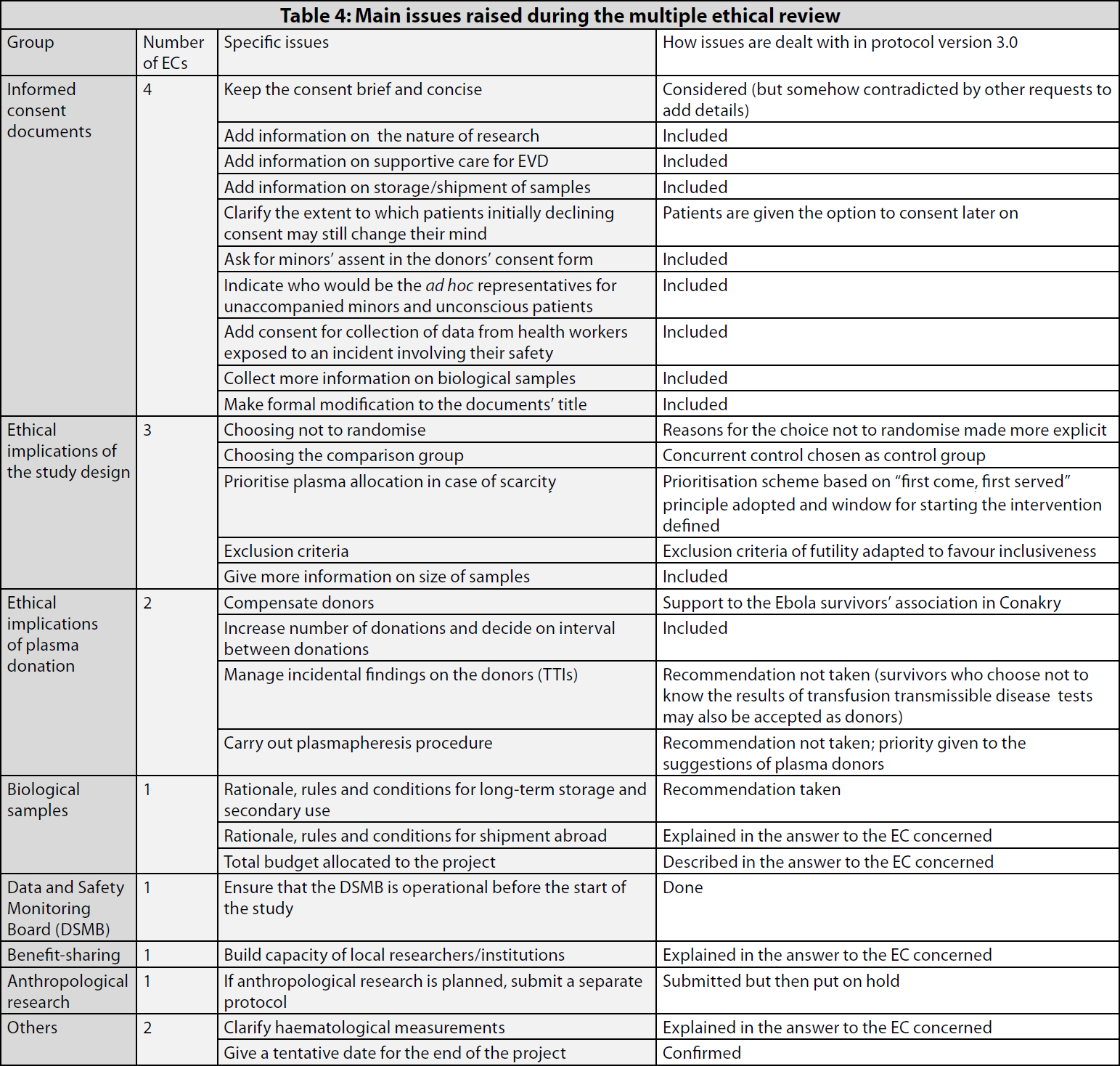
- First, explicit clarifications were sought on why a randomised design had not been considered at all (the non-randomised design may reduce the likelihood to obtain conclusive results).
- Second, clarifications were requested on how the comparison group would be identified.
- Third, there was a call for a clearer and more detailed procedure with respect to the criteria for the prioritisation of plasma allocation, in case of scarcity.2
- Fourth, clarifications were requested on the exclusion criteria, in particular, futility, with an underlying “push” to maximise inclusiveness.
- The compensation to the plasma donors
- The number of donations per donor
- The management of possible incidental findings and, in particular, the issue of if and how positive results for transfusion transmissible infections (TTI) would be disclosed to participants
- The rationale for the long-term storage and shipment abroad of biological samples, and the rules and conditions for any future use
- The total budget allocated to this project
- The measures taken for capacity-building of local researchers and partner institutions
Discussion
The experience of the Ebola-Tx showed that the double ethical review becomes more complex in the backdrop of a public health emergency. First, the complexity of research consortia and the consequently high number of ECs involved resulted in multiple, rather than double, ethical review. (The number of ECs reviewing the Ebola-Tx could have been even higher if all the partner institutions had decided to submit the protocol to their own EC/IRB.) Second, there were major differences in the ECspecific submission forms, and their completion, whether on paper or through the web, was somewhat complex and timeconsuming for the academic sponsor. Third, we had considered sending a common reply letter so that each EC would have a clear overview of everybody else’s replies. This option had to be dropped for practical reasons: we started working on each reply letter the day that the comments of a given EC were received, in an attempt to minimise the time needed for resubmission (the sense of urgency was somehow exacerbated by the fact that re-submission took place in the period between Christmas and New Year’s Eve). However, a common answer, possibly pooling all comments in an anonymised way (listing all the comments without specifying from which EC they came), would have allowed the ECs to get the overall picture in addition to viewing their own set of questions and answers. However, in practice, to our knowledge there was neither direct, nor indirect communication among the ECs involved in the review. Time-wise, all ECs did their best to ensure that the review was performed as soon as possible, often adapting their own procedures in accordance with the urgency of the situation. The total time to approval was 55 days. The ethical review, even if complex, was not the main hurdle that delayed the start of the study: the last approval was obtained on January 26, while the first plasma collection started on February 9 and the first CP administration was done on February 19. The case might be different for trials assessing investigational medicinal products that do not require the same complex technical set-up and the same level of engagement with a stigmatised and vulnerable group such as the Ebola survivors. The reviews varied greatly in terms of the format and the length of the answer, as well as content-wise. Some ECs sent reviews that were quite succinct, while others framed the required changes in a narrative text explaining the ethical reasoning behind their requests. ECs with greater experience in the review of research conducted in vulnerable communities and research-limited contexts provided more comprehensive and targeted comments, although the levels of detail differed. The inputs of the ethical review led to a clear improvement in some aspects of the trial. These include, for instance, the addition of consent for collecting data from health workers who would have been exposed to an incident involving their safety, and the replacement of a broad exclusion criterion of futility by more focused exclusion criteria, aiming at maximising inclusiveness. The critical question raised by various ECs as to who to treat in case of plasma scarcity resulted in a prioritisation scheme based on the “first come, first served” criterion. These changes also made the protocol more acceptable for the team on site. The comments concerning plasma donors, conversely, did not lead to substantial modifications of the protocol. In particular, the suggestion to exclude a priori those potential donors who were not willing to know the results of TTI tests was rejected after consultation with representatives of the donors themselves. The important issue raised by one EC on the shipment and long-term storage of samples abroad did not result in substantial changes to the protocol, but more details on this issue were added in the informed consent form, thus improving the information given to the participants. Also, the rationale for the proposed strategies became clearer, and researchers were encouraged to give attention to the issue of future biobanking throughout and after the study. The request for describing measures for building the capacity of the local researchers and partner institutions, also raised by just one EC, did not lead to modifications in the protocol. However, plans were described in the answer to the EC concerned, and long-term plans that went beyond the study period itself were designed. We have seen that in theory, all ECs preferred short and concise informed consent documents, but once all the comments were incorporated, the revised consent documents appeared to be longer than the original. This might have been avoided if a joint ethical review had been carried out, rather than assembling different comments from different sources. Overall, the variability observed across the reviews of the different ECs resulted in just a few contradictions, concerning the contents of the informed consent documents, and in a lot of complementarity. The different perspectives of the various ECs made it possible to cover a broader range of ethical issues, which had a positive influence on the ethical soundness of the study. These findings are in line with those of other groups. In particular, the 4ABC study group (13) found that the process of double ethical review led to important complementarities. This study on malaria was carried out in seven African countries and was also sponsored by the ITM. The Belgian ECs highlighted the ethical aspects related to indemnification for harm, insurance and confidentiality, while the African ECs focused on the need to ensure co-ownership of the study data, the sites’ qualification and capacity, the transfer of biological samples abroad, and the appropriateness of reimbursement of the patients’ travel expenses (4). These observations could appear to be in contrast with other groups’ findings concerning the multiple ethical review in multicentre trials. For instance, Burman and colleagues observed that the local approval of two multicentre clinical trials was time-consuming and resulted in many changes in the centrally approved consent forms, which often decreased readability and introduced errors (14). Silverman and colleagues noticed a high degree of variability among the research practices of the IRBs of the institutions participating in a multicentre trial (15). These discrepancies could indicate that the double (or multiple) ethical review is particularly useful for externally sponsored, North–South collaborative clinical research. It is in these contexts that, rather than resulting in a simple multiplication of approvals, the double ethical review is likely to improve the ethical soundness of research through the complementarity of perspectives from the country(ies) of the sponsor and the country(ies) of the study participants (1, 2). However, the lack of communication among all the ECs concerned may give rise to some contradictions in the reviews, and this regrettably results in a lack of mutual learning between the northern and southern ECs (4).Conclusion
Overall, the double ethical review improved the quality of the Ebola-Tx protocol and led to better protection of patients and the community, thanks to the complementarity of the different reviews. Our findings support the position that the double ethical review is beneficial in North–South collaborative clinical research, and that it should be routinely required and implemented in externally sponsored trials. The quality and coherence of the review would have been enhanced by joint ethical reviews, or at least by direct dialogue between the reviewing bodies, in accordance with the recommendations of the EVD WHO Background Document of September 2014 (“flexible approaches are required to harmonise various review processes, and ensure that the various ECs can review the projects simultaneously and share and discuss the review outcomes with each other”). However, the time constraint imposed by the epidemic did not allow for harmonisation of the various review processes, and there was no scope for the various ECs to share and discuss the review outcomes with each other. During public health emergencies, much more should be done to harmonise the review process, such as fostering direct dialogue among ECs. Joint ethical review would also be beneficial, but would be feasible only if ad hoc mechanisms were planned before the emergence of the next outbreak.
Acknowledgements
We are grateful to all the ethics committees and institutional review boards involved in reviewing the trial protocol for ensuring smooth collaboration, and to all the consortium partners and national and international stakeholders involved in the Ebola- Tx trial. We would also like to thank Annick Antierens, the MSF research coordinator who managed the submissions to the MSF ERB.
Funding
The Ebola-Tx project is funded by the European Union’s Horizon 2020 research and innovation programme under grant agreement No 666094. Additional funding is provided by the Department of Economy, Science and Innovation of the Flemish government. The funders have not been involved in the present assessment of the multiple ethical review.
Conflict of interest
All the authors are affiliated to research institutions which participated in the Ebola-Tx trial and are members of the Trial Management Group.
Raffaella Ravinetto is a member of the ERB of the MSF. She did not participate in the ethical review of the Ebola-Tx protocol.
Notes
1 For MSF ERB, the submission package was sent on this date to the MSF research coordinator 2 It is noteworthy that this remained a theoretical scenario since in practice, plasma was always available to all eligible patients during the study.References
- International Ethical Guidelines for Biomedical Research Involving Human Subjects. Council for International Organizations of Medical Sciences (CIOMS) in collaboration with the WHO. Geneva, Switzerland; 2002.
- Nuffield Council on Bioethics. The ethics of research related to healthcare in developing countries. London: Nuffield Council on Bioethics; 2002
- Bompart F, Hirsch F, Bertoye PH, Vray M. Bonnes Pratiques Cliniques dans les pays en développement: recommandations en termes d’application. Thérapie. 2008;63(2):77-82 (in French).
- Ravinetto R, Buve A, Halidou T, Lutumba P, Talisuna A Juffrie M, D’Alessandro U, Boelaert M. Double ethical review of North-South collaborative clinical research: hidden paternalism or real partnership? Trop Med Int Health. 2011;16(4):527-30. doi: 10.1111/j.1365- 3156.2011.02732.x. Epub 2011 Feb 1.
- FDRE Ministry of Science and Technology. National Research Ethics Review Guideline. Fifth edition, September 2014 [cited 2015 Sep 14]. Available from: http://www.ccghr.ca/wp-content/uploads/2013/11/national-research-ethics-review-guidline.pdf
- Uganda National Council for Sciences and Technology (UNCST). National Guidelines for Research Involving Humans as Research Participants. Kampala, Uganda: UNCST, July 2014 [cited 2015 Sep 14]. Available from: http://www.uncst.go.ug/dmdocuments/Human%20Subjects%20Protection%20Guidelines%20July%202014.pdf
- Baize S, Pannetier D, Oestereich L, Rieger T, Koivogui L, Magassouba N, Soropogui B, Sow MS, Keïta S, De Clerck H, Tiffany A, Dominguez G, Loua M, Traoré A, Kolié M, Malano ER, Heleze E, Bocquin A, Mély S, Raoul H, Caro V, Cadar D, Gabriel M, Pahlmann M, Tappe D, Schmidt-Chanasit J, Impouma B, Diallo AK, Formenty P, Van Herp M, Günther S. Emergence of Zaire Ebola virus disease in Guinea. N Engl J Med. 2014;371(15):1418-25. doi: 10.1056/NEJMoa1404505. Epub 2014 Apr 16.
- Coordination Nationale Ebola in Guinée, OMS., Rapport de la Situation Epidémiologique, Maladie à Virus Ebola en Guinée du 25 Juin 2015. Sit. Rep. No 436, 2015, Coordination Nationale Ebola Guinée and Organisation mondiale de la Santé: Conakry, Guinea [cited 2015 Sep 14]. Available from: http://guinea-ebov.github.io/
- World Health Organisation. Background document on potential Ebola therapies and vaccines, September 2014 [cited 2015 Sep 14]. Available from: http://www.who.int/mediacentre/news/statements/2014/ebolatherapies-consultation/en/
- World Health Organisation. Use of convalescent whole blood or plasma collected from patients recovered from Ebola virus disease for transfusion, as an empirical treatment during outbreaks. Geneva: WHO, 2014. WHO/HIS/SDS/2014.8 [cited 2015 Sep 14]. Available from: http://www.searo.who.int/entity/emerging_diseases/ebola/who_his_sds_2014.8_eng.pdf
- Schopper D, Dawson A, Upshur R, Ahmad A, Jesani A, Ravinetto R, Selgelid MJ, Sheel S, Singh J. Innovations in research ethics governance in humanitarian settings. BMC Med Ethics. 2015;16:10. doi: 10.1186/s12910-015-0002-3.
- Adebamowo C, Bah-Sow O, Binka F, Bruzzone R, Caplan A, Delfraissy JF, Heymann D, Horby P, Kaleebu P, Tamfum JJ, Olliaro P, Piot P, Tejan-Cole A, Tomori O, Toure A, Torreele E, Whitehead J. Randomised controlled trials for Ebola: practical and ethical issues. Lancet. 2014;384(9952):1423-4. doi: 10.1016/S0140-6736(14)61734-7. Epub 2014 Oct 13.
- Four Artemisinin-Based Combinations (4ABC) Study Group. A head-tohead comparison of four artemisinin-based combinations for treating uncomplicated malaria in African children: a randomized trial. PLoS Med. 2011;8(11):e1001119. doi: 10.1371/journal.pmed.1001119. Epub 2011 Nov 8.
- Burman W, Breese P, Weis S, Bock N, Bernardo J, Vernon A; Tuberculosis Trials Consortium. The effects of a local review on informed consent documents from a multicenter clinical trials consortium. Control Clin Trials. 2003;24(3):245-55.
- Silverman H, Hull SC, Sugarman J. Variability among institutional review boards’ decisions within the context of a multicenter trial. Crit Care Med. 2001;29(2):235-41.